| |||||||||||||
| |||||||||||||
| Ogólne informacje | |||||||||||||
| Wzór sumaryczny |
C19H25BN4O4 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Masa molowa |
384,24 g/mol | ||||||||||||
| Wygląd |
biały bezpostaciowy proszek[1] | ||||||||||||
| Identyfikacja | |||||||||||||
| Numer CAS | |||||||||||||
| PubChem | |||||||||||||
| DrugBank | |||||||||||||
| |||||||||||||
| |||||||||||||
| |||||||||||||
| |||||||||||||
| Jeżeli nie podano inaczej, dane dotyczą stanu standardowego (25 °C, 1000 hPa) | |||||||||||||
| Klasyfikacja medyczna | |||||||||||||
| ATC | |||||||||||||
| Stosowanie w ciąży |
kategoria D | ||||||||||||
| |||||||||||||
| |||||||||||||
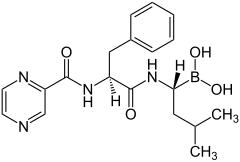
Bortezomib (łac. bortezomibum) – organiczny związek chemiczny, dwupeptydowa pochodna kwasu borowego, będąca silnym i odwracalnym inhibitorem proteasomu 26S[3] (kompleksu białkowego odpowiadającego za degradację białek komórkowych).
Bortezomib jest pierwszym i, jak dotąd, jedynym wprowadzonym do leczenia inhibitorem proteasomu[4][5]. W badaniach klinicznych II fazy stwierdzono istotną odpowiedź na leczenie u ponad 1/3 pacjentów z zaawansowanym szpiczakiem mnogim[6]. Na tej podstawie amerykańska Agencja Żywności i Leków w dniu 13 maja 2003 roku zarejestrowała bortezomib w trybie przyspieszonym ze wskazaniem leczenia szpiczaka mnogiego u chorych, u których po zastosowaniu co najmniej dwóch schematów leczenia nastąpił nawrót choroby[7]. Następnie Agencja Żywności i Leków zatwierdziła również leczenie chłoniaka z komórek płaszcza bortezomibem u pacjentów, którzy otrzymali wcześniej co najmniej jeden cykl leczenia[8].
Historia
Bortezomib został po raz pierwszy zsyntetyzowany w 1995 roku w zespole kierowanym przez J. Adamsa, w firmie Myogenics (następnie ProScript), jako wynik badań nad inhibicją proteasomu. Wybrano do tego celu związki zawierające bor (a konkretnie kwas borowy), gdyż cechowały się one najlepszymi właściwościami i wysoką swoistością. Ze wszystkich zsyntetyzowanych związków, do dalszych badań wybrano właśnie bortezomib[9]. Wyniki badań przedklinicznych były obiecujące, lek wszedł więc w pierwszą fazę badań klinicznych, gdzie oceniano jego skuteczność na małej grupie pacjentów ze szpiczakiem mnogim. Firma ProScript popadła w kłopoty finansowe i w maju 1999 roku została przejęta przez Leukosite. W październiku tego samego roku Leucosite kupiła firma Millenium Pharmaceuticals. Dalsze badania nad bortezomibem (oznaczonym symbolem MG-341, a następnie PS-341) zostały wstrzymane, ze względu na większe zainteresowanie macierzystymi projektami firmy. Sytuacja uległa zmianie, gdy okazało się, że u jednego z pacjentów uczestniczących w badaniach klinicznych I fazy uzyskano całkowitą odpowiedź na leczenie bortezomibem. Późniejsze wyniki innych badań wskazywały na to, że podobne efekty można osiągnąć u 15% pacjentów ze szpiczakiem mnogim leczonym bortezomibem[4].
Mechanizm działania
Funkcjonowanie szlaku ubikwityna-proteasom 26S
Za sprawną degradację białek komórkowych odpowiada szlak ubikwityna–proteasomy. Reguluje on istotne procesy w komórce, między innymi transdukcję sygnałów, regulację transkrypcji, odpowiedź na stres, a także kontrolę czynności receptorów[10]. Szlak ten pełni również kontrolę nad aktywacją czynnika transkrypcyjnego NF-κB, degradując inhibitor tego białka – I-κB[11]. Proteasom 26S jest białkiem o masie 1500–2000 kD, składającym się z kompleksu katalitycznego 20S (o masie około 700 kD) i kompleksu pełniącego rolę regulatorową – 19S[4][10][12]. Biorąc pod uwagę budowę przestrzenną, proteasom składa się z dwóch zewnętrznych i dwóch wewnętrznych pierścieni, które razem tworzą strukturę cylindryczną mającą trzy przedziały[13]. Obydwa pierścienie zewnętrzne mają po siedem podjednostek α każdy (α1-α7), a pierścienie wewnętrzne – siedem podjednostek β każdy (β1-β7). Kompleks katalityczny ma aktywność: chymotrypsyno-, trypsyno- i kaspazopodobną[12]; aktywne miejsca katalityczne znajdują się po wewnętrznej stronie cylindra[14]. Proces degradacji białek rozpoczyna się od przyłączenia do nich łańcucha poliubikwitynowego (tzw. ubikwitynacja). Tak znakowane białka rozpoznawane są przez kompleks regulatorowy, gdzie usunięty zostaje znacznik ubikwitynowy. Następnie ATP-azy o aktywności chaperonopodobnej rozwijają białka przeznaczone do degradacji i wprowadzają je do wnętrza kompleksu katalitycznego 20S[15]. Otwory te są bardzo małe (około 1,3 nm), dlatego konieczne jest znaczne rozwinięcie degradowanych białek[14]. Dodatkowo otwór wejściowy zablokowany jest „bramką” – N-końcowym fragmentem łańcucha podjednostki α3. Blokada jest jednak zwalniana, gdy podjednostki 20S i 19S są ze sobą związane[16]. Gdy białka przeznaczone do degradacji znajdą się we wnętrzu podjednostki katalitycznej, są hydrolizowane przez sześć miejsc aktywnych (po dwa na podjednostkach β1, β2 i β5). W ten sposób powstają ostateczne produkty hydrolizy – polipeptydy o długości 3–22 aminokwasów[4][14].
Mechanizm działania bortezomibu





Głównym działaniem bortezomibu jest blokowanie aktywności chymotrypsynopodobnej proteasomu. Zaobserwowano, że komórki nowotworowe inaczej reagują na jego inhibicję – najczęściej apoptozą – podczas gdy u zdrowych komórek następuje jedynie zahamowanie cyklu komórkowego[22]. Zmiany, jakie zachodzą w funkcjonowaniu komórek z zablokowanym proteasomem, można podzielić na trzy główne grupy[23]:
- zmiany związane z inhibicją NF-κB
- zmiany powiązane ze zwiększeniem aktywności szlaków proapoptotycznych
- zmiany w mikrośrodowisku nowotworu
NF-κB jest jednym z najważniejszych czynników transkrypcyjnych[24]. Reguluje on wzrost komórek, ewentualną ich apoptozę, a także czuwa nad ekspresją różnych cytokin, cząsteczek adhezyjnych i ich receptorów[12]. NF-κB znajduje się w cytoplazmie w stanie nieaktywnym – jest związany z inhibitorem I-κB. Pobudzenie komórek powoduje aktywację kaskad białkowych, które prowadzą do uczynnienia kinazy I-κB. Kinaza ta fosforyluje dwie reszty serynowe w N-końcowej domenie regulatorowej I-κB, dzięki czemu inhibitor NF-κB jest rozpoznawany przez ligazę ubikwityny E3 typu SCF i ulega ubikwitynacji, a to z kolei kieruje I-κB na drogę degradacji przez proteasomy[25]. Uwolniony NF-κB wędruje do jądra komórkowego, łączy się z regionami promotorowymi niektórych genów i aktywuje syntezę wielu cytokin i chemokin, cząsteczek adhezyjnych i cykliny D, gwarantujących komórce wzrost i przeżycie. Inhibicja proteasomów prowadzi do zahamowania aktywności NF-κB wskutek zablokowania degradacji I-κB[26]. Dochodzi do wzrostu stężenia białek proapoptotycznych, m.in. białka Bcl-2, co stymuluje uwalnianie cytochromu C, aktywuje kaskadę kaspaz i prowadzi do apoptozy[27]. Aktywowany NF-κB gra także dużą rolę w ekspresji komórkowych cząsteczek adhezyjnych, takich jak VCAM-1, przez które komórki szpiczaka mnogiego mogą wiązać się ze zrębem. Powoduje to zwiększenie wydzielania interleukiny-6 przez komórki zrębu, co hamuje apoptozę i odpowiada za oporność na chemioterapię[28]. Bortezomib, częściowo przez swój hamujący wpływ na NF-κB, zapobiega zwiększonej produkcji IL-6 i przyleganiu komórek szpiczaka do zrębu[29]. Zahamowanie wydzielania interleukiny-6 ma również znaczenie w blokowaniu unaczyniania nowotworu i jego wzrostu – wiadomo bowiem, że IL-6 należy do czynników wzrostu[30][31].
Nie do końca wiadomo, dlaczego komórki nowotworowe reagują apoptozą na zablokowanie czynności proteasomów. Wydaje się, że sama inhibicja NF-κB nie dość wyczerpująco opisuje skutki działania bortezomibu. Badania nad specyficznym inhibitorem kinazy I-κB – substancją o nazwie kodowej PS-1145 – wykazały jedynie 20–50% zahamowanie proliferacji komórek szpiczaka mnogiego przy stężeniu <12,5 μmol/l. Bortezomib zaś, przy stężeniu ≤0,1 μmol/l, hamował ją całkowicie[32]. W odróżnieniu od PS-1145 bortezomib indukuje również apoptozę komórek szpiczaka mnogiego[26][33].
Badania molekularne, wykorzystujące technologię mikromacierzy, wskazują na to, że bortezomib powoduje zwiększoną ekspresję genów białek szoku termicznego i białek proapoptotycznych, przy czym geny białek antyapoptotycznych i geny wzrostu ulegają znacznej supresji[27]. Bortezomib aktywuje kinazę JNK (kinazę N-końca białka c-jun), co prowadzi do ekspresji receptora śmierci dla cytokin – Fas (prawdopodobnie w wyniku zwiększenia ekspresji genu białka c-myc)[34][35] i aktywuje kaspazę-8 i kaspazę-3. Kaspaza-8 odpowiedzialna jest za mediowanie szlaku apoptozy niezależnie od apoptozy „kierowanej” przez kaspazę-9 (która ma związek z opisaną wcześniej inhibicją NF-κB). Wydaje się, że to właśnie aktywacja kinazy JNK jest ważnym szlakiem dla wywoływanej przez bortezomib apoptozy komórek szpiczaka mnogiego[27]. Uczynnienie kaspazy-3 prowadzi do fosforylacji białka p53 (seryna w pozycji 15), zwiększając przez to aktywność jego dzikiej formy, co prowadzi do apoptozy w wyniku wzrostu stężenia białka Bax i p21[36].
Bortezomib wywołuje również zakłócenia w przekazywaniu sygnałów w komórce. Zwiększa się stężenie białek p21, p27 (będących inhibitorami cyklinozależnych kinaz)[37], p15, p16, p18, p19 (inhibitory kinazy 4/6 zależnej od cykliny D)[38], co zatrzymuje cykl komórkowy w fazie G1/S i wywołuje apoptozę. Zwiększone stężenie białka Bax przeważa nad nadmierną ekspresją białka Bcl-2 i również prowadzi do programowanej śmierci komórki[39]. Uszkodzone białka nie mogą zostać rozłożone przez zablokowany proteasom[40], w komórce pojawiają się więc sygnały proapoptotyczne przy jednoczesnych sygnałach antyapoptotycznych. Ta sprzeczność sygnałów w komórce jest również wynikiem nieuporządkowanych zmian w stężeniach cyklin A, B, C i D, co w połączeniu ze zwiększonym stężeniem inhibitorów kinaz zależnych od cyklin sprzyja apoptozie. Podejrzewa się, że współistnienie w komórce sygnałów pro- i antyapoptotycznych prowadzi komórkę ku apoptozie[41].
Bortezomib powoduje również zmniejszenie ekspresji insulinopodobnego czynnika wzrostu-1 i odpowiedniego receptora[27], zahamowanie szlaku Ras/Raf/kinaza białkowa aktywowanego interleukiną-6 i mitogenami[26] oraz zwiększenie ilości białek szoku cieplnego[42] (przede wszystkim hsp90[43]).
Bortezomib jest związkiem wysoce selektywnym. Przy stężeniu 10 μmoli nie hamuje żadnego z wielu przebadanych receptorów i enzymów. Jest jednocześnie około 1500 razy bardziej selektywny w stosunku do proteasomu w porównaniu do następnego preferowanego enzymu. Inhibicja proteasomu przez bortezomib jest odwracalna, a jej okres półtrwania wynosi 20 minut[22].

Badania kliniczne
W jednym z badań klinicznych I fazy uzyskano całkowitą odpowiedź na leczenie bortezomibem u jednego chorego na szpiczaka mnogiego, co stwierdzono na podstawie ujemnego wyniku immunofiksacji, oraz osiem przypadków znaczącego zmniejszenia stężenie białka monoklonalnego w osoczu i nacieków nowotworowych w szpiku kostnym. Uzyskano również dobre wyniki w przypadku chorego cierpiącego na chłoniaka nieziarniczego[45]. W innym badaniu pierwszej fazy ocenie poddano skuteczność bortezomibu w przypadku chorych z zaawansowaną postacią guzów litych, jak niedrobnokomórkowy rak oskrzela, różne rodzaje raka nosogardła, czerniak i rak nerki[46]. W II fazie badań klinicznych, prowadzonej przez zespół Richardsona, oceniano skuteczność bortezomibu w przypadku chorych z nawrotem szpiczaka mnogiego i ze szpiczakiem mnogim opornym na leczenie[6]. Terapia składała się z 8 cykli, a każdy cykl obejmował podawanie bortezomibu w dawce 1,3 mg/m² powierzchni ciała, dwa razy w tygodniu, z tygodniem bez leczenia. U pacjentów z odpowiedzią suboptymalną podawano deksametazon po dwóch pierwszych cyklach leczenia bortezomibem. Przed rozpoczęciem leczenia chorzy (202 pacjentów biorących udział w badaniu) otrzymywali kortykosterydy (100%), leki alkilujące (92%), antracykliny (81%), talidomid (83%), a 64% pacjentom przeszczepiono komórki macierzyste.
Spośród 193 chorych (9 pacjentów zmarło) u 4% uzyskano całkowitą odpowiedź na leczenie, u 6% – odpowiedź prawie całkowitą, u 18% – odpowiedź częściową, u 7% – odpowiedź minimalną. Całkowity odsetek pacjentów, którzy zareagowali na leczenie, wyniósł 35%. Wykazano również, że u 18% chorych opornych na leczenie deksametazonem uzyskano zwiększenie skuteczności leczenia tym lekiem, co może świadczyć o tym, że bortezomib znosi częściowo tę oporność[47]. W innym badaniu II fazy, prowadzonym przez zespół Jagannatha, badano rezultaty leczenia bortezomibem u 54 pacjentów chorujących na szpiczaka mnogiego, którzy mieli nawrót choroby lub wykazywali oporność na terapię pierwszego rzutu. Chorych podzielono na dwie grupy, a następnie podawano im bortezomib w dawce 1,0 lub 1,3 mg/m² powierzchni ciała, dwa razy w tygodniu przez 2 tygodnie, co 3 tygodnie, przez maksymalnie 8 cykli. Deksametazon włączano do leczenia u osób z chorobą stabilną/postępującą po odpowiednio 4 lub 2 cyklach. Całkowitą odpowiedź uzyskano u 2 pacjentów, a częściową u 8. W połączeniu z deksametazonem, bortezomib wykazywał skuteczność rzędu 50% (jako suma odpowiedzi całkowitych i częściowych). Randomizowane, wieloośrodkowe badanie APEX (assessment of proteasome inhibition for extending remissions) wykazało większą skuteczność bortezomibu stosowanego w monoterapii w porównaniu z deksametazonem w dużych dawkach[48].
W prospektywnych, międzynarodowych, randomizowanych, otwartych badaniach III fazy badano skuteczność bortezomibu zarówno w przypadkach wcześniej nieleczonego szpiczaka mnogiego, jak i szpiczaka mnogiego nawracającego lub opornego na leczenie. W pierwszym przypadku badaniem VISTA objęto 682 pacjentów i miało ono na celu określenie, czy podawanie bortezomibu (1,3 mg/m²), w skojarzeniu z melfalanem (9 mg/m²) i prednizonem (60 mg/m²), pacjentom z wcześniej nieleczonym szpiczakiem mnogim prowadzi do poprawy tzw. wskaźnika TTP („czas do progresji choroby”), w porównaniu z leczeniem bez bortezomibu. Okres terapii wynosił maksymalnie 9 cykli, czyli około 54 tygodni. Wykazano zwiększenie mediany przeżycia do ponad 20 miesięcy w porównaniu z 15 miesiącami w przypadku programu leczenia bez bortezomibu. Zwiększył się również odsetek odpowiedzi na leczenie – z 35% w przypadku leczenia jedynie melfalanem i prednizonem do 71% w przypadku zastosowania bortezomibu[49][50].
W drugim przypadku, skuteczność bortezomibu w porównaniu z deksametazonem oceniano w dwóch badaniach klinicznych III fazy, w których podawano bortezomib w standardowej dawce 1,3 mg/m². Obejmowało ono 669 pacjentów z nawrotowym i opornym na leczenie szpiczakiem mnogim, którzy wcześniej byli leczeni za pomocą od 1 do 3 programów terapeutycznych lub którzy leczeni byli za pomocą przynajmniej dwóch programów terapii i u których podczas stosowania ostatniego z nich nastąpiła progresja choroby. Badania wykazały, że stosowanie bortezomibu znamiennie wydłuża medianę przeżycia z ponad 8 miesięcy (deksametazon) do 17 miesięcy (bortezomib). Odsetek osób, które odpowiedziały na leczenie również poprawił się znamiennie – z 18% (deksametazon) do 38% (bortezomib). Całkowitą odpowiedź na leczenie zanotowano u 6% w porównaniu z <1% w przypadku deksametazonu[51].
Obecnie trwają wieloośrodkowe badania mające na celu ocenę skuteczności bortezomibu, zarówno w monoterapii, jak i w leczeniu skojarzonym w przypadkach innych chorób, między innymi:
- w połączeniu z metotreksatem i takrolimusem w chorobie przeszczep przeciw gospodarzowi[52]
- łącznie z rytuksymabem i deksametazonem w makroglobulinemii Waldenströma[53].
Synteza chemiczna
Pierwszym etapem syntezy bortezomibu jest kondensacja pochodnej kwasu aminoboronowego, w trakcie której atom boru zabezpieczony jest cyklicznym estrem pinanodiolu, z fenyloalaniną zablokowaną grupą butyloksykarbonylową. Następnie usuwa się zabezpieczenie z N-końca i tak otrzymany związek poddaje się kolejnej kondensacji z kwasem pirazynokarboksylowym. Otrzymany produkt poddaje się reakcji z kwasem izobutyloboronowym, który usuwa pozostałe zabezpieczenie przy atomie boru. Końcowym produktem reakcji jest bortezomib[54].

Farmakokinetyka
Bortezomib podany dożylnie w postaci szybkiego bolusa szybko osiąga maksymalne stężenie we krwi. Duża objętość dystrybucji świadczy o znacznej dystrybucji do tkanek obwodowych. Bortezomib łączy się z białkami osocza średnio w 83%. Wartość ta nie jest zależna od stężenia osiąganego w surowicy. Bortezomib jest metabolizowany w wątrobie, przy udziale izoenzymów cytochromu P-450 – CYP3A4, CYP2C19 i CYP1A2. Główny szlak metaboliczny polega na deboronacji do dwóch nieczynnych metabolitów, które następnie podlegają różnorodnej hydroksylacji. Średni czas półtrwania bortezomibu w osoczu waha się w granicach 40–193 godzin w przypadku dawek wielokrotnych. Przy dawce jednorazowej, połowiczny czas trwania jest krótszy i wynosi 4–15 godzin. Drogi eliminacji bortezomibu nie zostały zbadane[55].
Wskazania
Bortezomib jest wskazany:
- w leczeniu skojarzonym z melfalanem i prednizonem albo/oraz z przeszczepieniem szpiku kostnego u chorych z wcześniej nieleczonym szpiczakiem mnogim, którzy nie kwalifikują się do chemioterapii dużymi dawkami cytostatyków
- w monoterapii u pacjentów z progresją szpiczaka mnogiego, którzy wcześniej otrzymali co najmniej jeden program leczenia oraz u których przeszczepiono już szpik lub też do takiego leczenie nie się kwalifikują[56].
Przeciwwskazania
- nadwrażliwość na bortezomib, związki boru lub jakikolwiek inny składnik preparatu
- ciężka niewydolność wątroby
- ostra rozlana naciekowa choroba płuc i osierdzia
Ostrzeżenia specjalne
- Leczenie bortezomibem często wywołuje objawy uboczne ze strony przewodu pokarmowego, w tym niedrożność jelit. Z tego względu pacjenci cierpiący na zaparcia powinni być uważnie monitorowani.
- Najczęstszym działaniem niepożądanym hematologicznym jest małopłytkowość. Dlatego też liczba płytek krwi powinna być sprawdzana przed każdym podaniem bortezomibu. W przypadku spadku ich poziomu poniżej 25000/μl należy wstrzymać podawanie leku. Może ono zostać wznowione, z zastosowaniem zmniejszonych dawek, dopiero po uzyskaniu poprawy w obrazie morfologicznym krwi[57].
- Podczas leczenia bortezomibem bardzo często pojawia się neuropatia obwodowa (głównie czuciowa). Zanotowano również kilka przypadków ciężkiej neuropatii ruchowej. Pacjentów należy bacznie obserwować w kierunku wystąpienia objawów neuropatii, takich jak: uczucie pieczenia, hiperestezja, hipestezja, parestezja, ból neuropatyczny lub osłabienie[58].
- Należy zwrócić szczególną uwagę na pacjentów z padaczką lub tych, u których występują czynniki ryzyka wystąpienia drgawek.
- Leczeniu bortezomibem często towarzyszy niedociśnienie ortostatyczne/niedociśnienie zależne od pozycji ciała. Może to być związane z neuropatią układu autonomicznego wywołaną przez podawanie bortezomibu lub też nasiloną przez lek. Należy zachować ostrożność w przypadku pacjentów przyjmujących leki mogące wywołać niedociśnienie oraz w przypadku pacjentów odwodnionych. Chorych leczonych bortezomibem należy poinformować, by zgłosili się do lekarza, gdy zauważą u siebie takie objawy, jak: zawroty głowy, uczucie pustki w głowie i omdlenia[59].
- Pacjenci z chorobami serca lub u których występują czynniki ryzyka jej rozwoju, powinni być ściśle monitorowani ze względu na to, że leczenie bortezomibem może sprzyjać jej wystąpieniu lub też zaostrzać objawy już istniejącej choroby serca. Tyczy się to w szczególności chorych z zastoinową niewydolnością serca[60].
- Zanotowano pojedyncze przypadki wydłużenia odstępu QT, nie ustalono jednak związku przyczynowego z leczeniem bortezomibem.
- Zaleca się wykonanie zdjęcia rentgenowskiego klatki piersiowej przed rozpoczęciem leczenia bortezomibem w celu określenia ewentualnego ryzyka wystąpienia zapalenia płuc, śródmiąższowego zapalenia płuc, nacieków w płucach o nieznanej etiologii oraz zespołu ostrej niewydolności oddechowej. Podczas prób klinicznych zanotowano kilka przypadków ostrych chorób płuc o nieznanej przyczynie, w niektórych przypadkach zakończone zgonem pacjenta[61].
- Pacjenci z niewydolnością wątroby lub nerek[62] powinni być bacznie obserwowani. Nie zaleca się leczenia bortezomibem u chorych cierpiących na ciężką postać niewydolności wątroby.
- Należy uważnie obserwować pacjentów, u których przed rozpoczęciem leczenia masa nowotwory była duża, ze względu na wystąpienie zespołu rozpadu guza[63].
- Należy zachować ostrożność wobec pacjentów cierpiących na choroby związane z gromadzeniem się białka (np. amyloidoza)[64].
- Chorzy przyjmujący bortezomib nie powinni prowadzić pojazdów mechanicznych ani obsługiwać maszyn.
Interakcje
Należy zachować szczególną ostrożność przy jednoczesnym podawaniu bortezomibu z:
- silnymi inhibitorami CYP3A4, między innymi ketokonazolem, rytonawirem
- fluoksetyną oraz innymi lekami, będącymi inhibitorami CYP2C19[65]
- silnymi induktorami CYP3A4, na przykład ryfampicyną.
Zielona herbata (konkretnie galusan epigallokatechiny)[66][67] oraz kwas askorbinowy[68] niemal całkowicie hamują działanie bortezomibu.
Działania niepożądane
Do najczęściej występujących skutków ubocznych wynikających z leczenia bortezomibem należą:
- zakażenia wirusem półpaśca (w tym postać rozsiana)[69]
- hematotoksyczność (małopłytkowość, neutropenia, niedokrwistość)
- utrata apetytu
- zaburzenia neurologiczne (neuropatia obwodowa, obwodowa neuropatia czuciowa, parestezje, bóle głowy)[70]
- duszności
- zaburzenia ze strony układu pokarmowego (wymioty, biegunka, nudności, zaparcia)[71]
- wysypka
- bóle mięśni
- zmęczenie
- gorączka.
Do rzadziej występujących działań niepożądanych zaliczyć można:
- zakażenia górnych dróg oddechowych, zakażenia wirusem opryszczki[72]
- leukopenię, limfopenię
- odwodnienie
- hipokaliemię, hipoglikemię
- stany dezorientacji, depresję, bezsenność, lęk
- polineuropatię, zaburzenia smaku i czucia, niedoczulicę, drżenie
- zaburzenia widzenia
- zawroty głowy
- niedociśnienie ortostatyczne/niedociśnienie związane z pozycją ciała,
- zapalenie żył w miejscu wstrzyknięcia
- krwawienia z nosa, katar
- kaszel, duszność wysiłkową
- zaburzenia dyspeptyczne (wzdęcia, niestrawność, bóle brzucha)
- suchość w ustach
- bóle gardła i krtani
- obrzęki w okolicach oczu
- pokrzywkę, świąd, rumień, wypryski[73]
- zwiększoną potliwość
- złe samopoczucie i objawy grypopodobne
- zmniejszenie masy ciała
- zwiększenie aktywności dehydrogenazy mleczanowej we krwi.
Rzadko mogą wystąpić:
- różne zakażenia wywołane przez bakterie[74], wirusy lub grzyby chorobotwórcze
- zespół rozpadu guza
- pancytopenia, niedokrwistość hemolityczna, plamica małopłytkowa, limfadenopatia
- nadwrażliwość na lek
- zaburzenia w wydzielaniu hormonu antydiuretycznego
- zaburzenia gospodarki mineralnej
- zaburzenia psychiczne
- porażenie poprzeczne
- krwotok wewnątrzczaszkowy
- krwotok podpajęczynówkowy
- drgawki
- zespół niespokojnych nóg
- zaburzenia słuchu
- zaburzenia w pracy serca i układu krążenia
- zaburzenia wątroby i nerek
- zaburzenia erekcji, ból jąder.
Dawkowanie[75]
Dawkowanie w monoterapii
Początkowa dawka bortezomibu powinna wynosić 1,3 mg/m² powierzchni ciała, dwa razy w tygodniu, przez dwa tygodnie (w dniach 1, 4, 8 i 11), a następnie 10-dniowy okres odpoczynku (od 12 do 21 dnia). Opisany trzytygodniowy okres stanowi jeden cykl leczenia. Pomiędzy kolejnymi dawkami bortezomibu powinny upłynąć co najmniej 72 godziny. Zaleca się, by pacjenci, u których uzyskano odpowiedź całkowitą, otrzymali dodatkowe dwa cykle leczenia. Ponadto zaleca się, by pacjenci, którzy na leczenie odpowiedzieli częściowo, otrzymali w sumie 8 cykli. Dawkowanie powinno zostać dostosowane w razie wystąpienia neuropatii związanej z bortezomibem, w zależności od stopnia ciężkości tego działania niepożądanego[76]:
- neuropatia I. stopnia (parestezje, osłabienie i/lub zniesienie odruchów) bez występowania utraty funkcji – nie ma konieczności modyfikacji dawki
- neuropatia I. stopnia z bólem lub II. stopnia (z zaburzeniami funkcji, lecz nie utrudniający codziennej aktywności) – dawkę należy zredukować do 1,0 mg/m² pc.
- neuropatia II. stopnia z bólem lub III. stopnia (z utrudnieniami w codziennej aktywności) – należy przerwać podawanie bortezomibu do momentu ustąpienia objawów toksycznych. Następnie leczenie należy rozpocząć od zredukowanej dawki 0,7 mg/m² pc., podawanej tylko raz w tygodniu
- neuropatia IV. stopnia (neuropatia czuciowa, która utrudnia funkcjonowanie lub neuropatia ruchowa zagrażająca życiu lub prowadząca do porażenia) i/lub ciężka neuropatia autonomicznego układu nerwowego – leczenie bortezomibem należy przerwać
Dawkowanie w leczeniu skojarzonym
Bortezomib podawany jest razem z melfalanem i prednizonem przez dziewięć 6-tygodniowych cykli. W trakcie cykli 1–4 bortezomib podaje się dwa razy w tygodniu (dni: 1, 4, 8, 11, 22, 25, 29 i 32). W trakcie cykli 5–9 lek podaje się raz w tygodniu (dni: 1, 8, 22 i 29). Obowiązuje dawkowanie standardowe, czyli 1,3 mg/m² pc. Przed rozpoczęciem każdego cyklu należy określić liczbę płytek krwi i neutrofili oraz ocenić toksyczność niehematologiczną. Gdy liczba płytek wynosi ≥70·109/l, bezwzględna liczba neutrofili ≥1,0·109/l, a toksyczność niehematologiczna nie przekracza stopnia pierwszego, można wtedy rozpocząć nowy cykl leczenia. Gdy przekracza ona 3. stopień, leczenie bortezomibem należy przerwać, aż do momentu ustąpienia objawów toksycznych. Następnie dawkę należy zmniejszyć o jeden poziom (z 1,3 mg/m² na 1,0 mg/m² lub z 1,0 mg/m² na 0,7 mg/m²). Rozcieńczony roztwór bortezomibu podaje się dożylnie w formie bolusa, trwającego 3–5 sekund, do żył obwodowych lub przez centralny dostęp żylny. Po podaniu leku wkłucie powinno zostać przepłukane 0,9% roztworem chlorku sodu.
Preparaty
- Velcade (Janssen-Cilag) – fiolka zawierająca 1 mg lub 3,5 mg bortezomibu w postaci estru z mannitolem
Przypisy
- ↑ Vladimir Beljanski: Bortezomib. W: xPharm: The Comprehensive Pharmacology Reference. Elsevier, 2008. DOI: 10.1016/B978-008055232-3.63726-2.
- ↑ Bortezomib, [w:] DrugBank, University of Alberta, DB00188 (ang.).
- ↑ J.J. Shah, R.Z. Orlowski. Proteasome inhibitors in the treatment of multiple myeloma. „Leukemia”. 23 (11), s. 1964–1979, Nov 2009. DOI: 10.1038/leu.2009.173. PMID: 19741722.
- 1 2 3 4 R. Bold. Development of the proteasome inhibitor Velcade (Bortezomib) by Julian Adams, Ph. D., and Michael Kauffman, M.D., Ph. D. „Cancer Invest”. 22 (2), s. 328–329, 2004. PMID: 15199617.
- ↑ B.D. Cheson. Hematologic malignancies: new developments and future treatments. „Semin Oncol”. 29 (4 Suppl 13), s. 33–45, Aug 2002. PMID: 12170431.
- 1 2 S. Jagannath, B. Barlogie, J.R. Berenson, D.S. Siegel i inni. Updated survival analyses after prolonged follow-up of the phase 2, multicenter CREST study of bortezomib in relapsed or refractory multiple myeloma. „Br J Haematol”. 143 (4), s. 537–540, Nov 2008. DOI: 10.1111/j.1365-2141.2008.07359.x. PMID: 18783399.
- ↑ FDA Approval Letter for Velcade (bortezomib). [dostęp 2010-01-13].
- ↑ FDA approves bortezomib (Velcade) for the treatment of patients with mantle cell lymphoma who have received at least one prior therapy. [dostęp 2010-02-08]. [zarchiwizowane z tego adresu (2011-09-29)].
- ↑ J. Adams, V.J. Palombella, E.A. Sausville, J. Johnson i inni. Proteasome inhibitors: a novel class of potent and effective antitumor agents. „Cancer Res”. 59 (11), s. 2615–2622, Jun 1999. PMID: 10363983.
- 1 2 J. Adams, V.J. Palombella, P.J. Elliott. Proteasome inhibition: a new strategy in cancer treatment. „Invest New Drugs”. 18 (2), s. 109–121, May 2000. PMID: 10857991.
- ↑ V.J. Palombella, E.M. Conner, J.W. Fuseler, A. Destree i inni. Role of the proteasome and NF-kappaB in streptococcal cell wall-induced polyarthritis. „Proc Natl Acad Sci U S A”. 95 (26), s. 15671–15676, Dec 1998. PMID: 9861028.
- 1 2 3 J.B. Almond, G.M. Cohen. The proteasome: a novel target for cancer chemotherapy. „Leukemia”. 16 (4), s. 433–443, Apr 2002. DOI: 10.1038/sj.leu.2402417. PMID: 11960320.
- ↑ J. Löwe, D. Stock, B. Jap, P. Zwickl i inni. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. „Science”. 268 (5210), s. 533–539, Apr 1995. PMID: 7725097.
- 1 2 3 C.M. Pickart, A.P. VanDemark. Opening doors into the proteasome. „Nat Struct Biol”. 7 (11), s. 999–1001, Nov 2000. DOI: 10.1038/81018. PMID: 11062549.
- ↑ B.C. Braun, M. Glickman, R. Kraft, B. Dahlmann i inni. The base of the proteasome regulatory particle exhibits chaperone-like activity. „Nat Cell Biol”. 1 (4), s. 221–226, Aug 1999. DOI: 10.1038/12043. PMID: 10559920.
- ↑ M. Groll, M. Bajorek, A. Köhler, L. Moroder i inni. A gated channel into the proteasome core particle. „Nat Struct Biol”. 7 (11), s. 1062–1067, Nov 2000. DOI: 10.1038/80992. PMID: 11062564.
- ↑ J.H. Wang, R.B. Pepinsky, T. Stehle, J.H. Liu i inni. The crystal structure of an N-terminal two-domain fragment of vascular cell adhesion molecule 1 (VCAM-1): a cyclic peptide based on the domain 1 C-D loop can inhibit VCAM-1-alpha 4 integrin interaction. „Proc Natl Acad Sci U S A”. 92 (12), s. 5714–5718, Jun 1995. PMID: 7539925.
- ↑ B. Huang, M. Eberstadt, E.T. Olejniczak, R.P. Meadows i inni. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. „Nature”. 384 (6610). s. 638–641. DOI: 10.1038/384638a0. PMID: 8967952.
- ↑ Petros AM., Medek A., Nettesheim DG., Kim DH., Yoon HS., Swift K., Matayoshi ED., Oltersdorf T., Fesik SW. Solution structure of the antiapoptotic protein bcl-2. „Proceedings of the National Academy of Sciences of the United States of America”. 6 (98), s. 3012–3017, marzec 2001. DOI: 10.1073/pnas.041619798. PMID: 11248023.
- ↑ Riedl SJ., Li W., Chao Y., Schwarzenbacher R., Shi Y. Structure of the apoptotic protease-activating factor 1 bound to ADP. „Nature”. 7035 (434), s. 926–933, kwiecień 2005. DOI: 10.1038/nature03465. PMID: 15829969.
- ↑ RCSB PDB: Structure Summary. [dostęp 2010-01-14].
- 1 2 Jan Kazimierz (-1997) Podlewski, Alicja Chwalibogowska-Podlewska, Robert Adamowicz: Leki współczesnej terapii. Warszawa: Split Trading, 2005, s. 87–88. ISBN 83-85632-82-4.
- ↑ T. Hideshima, D. Chauhan, K. Podar, R.L. Schlossman i inni. Novel therapies targeting the myeloma cell and its bone marrow microenvironment. „Semin Oncol”. 28 (6), s. 607–612, Dec 2001. PMID: 11740818.
- ↑ N. Mitsiades, C.S. Mitsiades, V. Poulaki, D. Chauhan i inni. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. „Blood”. 99 (11), s. 4079–4086, Jun 2002. PMID: 12010810.
- ↑ M. Karin, M. Delhase. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. „Semin Immunol”. 12 (1), s. 85–98, Feb 2000. DOI: 10.1006/smim.2000.0210. PMID: 10723801.
- 1 2 3 T. Hideshima, D. Chauhan, P. Richardson, C. Mitsiades i inni. NF-kappa B as a therapeutic target in multiple myeloma. „J Biol Chem”. 277 (19), s. 16639–16647, May 2002. DOI: M200360200 10.1074/jbc. M200360200. PMID: 11872748.
- 1 2 3 4 N. Mitsiades, C.S. Mitsiades, V. Poulaki, D. Chauhan i inni. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. „Proc Natl Acad Sci U S A”. 99 (22), s. 14374–14379, Oct 2002. DOI: 10.1073/pnas.202445099. PMID: 12391322.
- ↑ L.A. Hazlehurst, W.S. Dalton. Mechanisms associated with cell adhesion mediated drug resistance (CAM-DR) in hematopoietic malignancies. „Cancer Metastasis Rev”. 20 (1–2), s. 43–50, 2001. PMID: 11831646.
- ↑ T. Hideshima, P. Richardson, D. Chauhan, V.J. Palombella i inni. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. „Cancer Res”. 61 (7), s. 3071–3076, Apr 2001. PMID: 11306489.
- ↑ R. LeBlanc, L.P. Catley, T. Hideshima, S. Lentzsch i inni. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. „Cancer Res”. 62 (17), s. 4996–5000, Sep 2002. PMID: 12208752.
- ↑ R.Z. Orlowski, P.M. Voorhees, R.A. Garcia, M.D. Hall i inni. Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. „Blood”. 105 (8), s. 3058–3065, Apr 2005. DOI: 10.1182/blood-2004-07-2911. PMID: 15626743.
- ↑ O. Fuchs, D. Provaznikova, I. Marinov, K. Kuzelova i inni. Antiproliferative and proapoptotic effects of proteasome inhibitors and their combination with histone deacetylase inhibitors on leukemia cells. „Cardiovasc Hematol Disord Drug Targets”. 9 (1), s. 62–77, Mar 2009. PMID: 19275578.
- ↑ S. Chu, M. Alexiadis, P.J. Fuller. Proteasome inhibition by bortezomib decreases proliferation and increases apoptosis in ovarian granulosa cell tumors. „Reprod Sci”. 16 (4), s. 397–407, Apr 2009. DOI: 10.1177/1933719108327589. PMID: 19087975.
- ↑ M. Berg, A. Lundqvist, P. McCoy, L. Samsel i inni. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. „Cytotherapy”. 11 (3), s. 341–355, 2009. DOI: 10.1080/14653240902807034. PMID: 19308771.
- ↑ M. Reis-Sobreiro, C. Gajate, F. Mollinedo. Involvement of mitochondria and recruitment of Fas/CD95 signaling in lipid rafts in resveratrol-mediated antimyeloma and antileukemia actions. „Oncogene”. 28 (36), s. 3221–3234, Sep 2009. DOI: 10.1038/onc.2009.183. PMID: 19561642.
- ↑ S.A. Vaziri, D.R. Grabowski, J. Hill, L.R. Rybicki i inni. Inhibition of proteasome activity by bortezomib in renal cancer cells is p53 dependent and VHL independent. „Anticancer Res”. 29 (8), s. 2961–2969, Aug 2009. PMID: 19661301.
- ↑ Y. Wang, A.K. Rishi, V.T. Puliyappadamba, S. Sharma i inni. Targeted proteasome inhibition by Velcade induces apoptosis in human mesothelioma and breast cancer cell lines. „Cancer Chemother Pharmacol”, Dec 2009. DOI: 10.1007/s00280-009-1181-8. PMID: 19960346.
- ↑ H. Holland, R. Koschny, W. Krupp, J. Meixensberger i inni. Cytogenetic and molecular biological characterization of an adult medulloblastoma. „Cancer Genet Cytogenet”. 178 (2), s. 104–113, Oct 2007. DOI: 10.1016/j.cancergencyto.2007.06.005. PMID: 17954265.
- ↑ G.B. Lesinski, E.T. Raig, K. Guenterberg, L. Brown i inni. IFN-alpha and bortezomib overcome Bcl-2 and Mcl-1 overexpression in melanoma cells by stimulating the extrinsic pathway of apoptosis. „Cancer Res”. 68 (20), s. 8351–8360, Oct 2008. DOI: 10.1158/0008-5472.CAN-08-0426. PMID: 18922907.
- ↑ T. Jung, T. Grune. The proteasome and its role in the degradation of oxidized proteins. „IUBMB Life”. 60 (11), s. 743–752, Nov 2008. DOI: 10.1002/iub.114. PMID: 18636510.
- ↑ H. Dong, L. Chen, X. Chen, H. Gu i inni. Dysregulation of unfolded protein response partially underlies proapoptotic activity of bortezomib in multiple myeloma cells. „Leuk Lymphoma”. 50 (6), s. 974–984, Jun 2009. DOI: 10.1080/10428190902895780. PMID: 19391038.
- ↑ A. Szabolcs, G. Biczó, Z. Rakonczay, L. Tiszlavicz i inni. Simultaneous proteosome inhibition and heat shock protein induction by bortezomib is beneficial in experimental pancreatitis. „Eur J Pharmacol”. 616 (1–3), s. 270–274, Aug 2009. DOI: 10.1016/j.ejphar.2009.05.019. PMID: 19486901.
- ↑ C. Fionda, A. Soriani, G. Malgarini, M.L. Iannitto i inni. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. „J Immunol”. 183 (7), s. 4385–4394, Oct 2009. DOI: 10.4049/jimmunol.0901797. PMID: 19748980.
- ↑ B.S. Moore, A.S. Eustáquio, R.P. McGlinchey. Advances in and applications of proteasome inhibitors. „Curr Opin Chem Biol”. 12 (4), s. 434–440, Aug 2008. DOI: 10.1016/j.cbpa.2008.06.033. PMID: 18656549.
- ↑ R.Z. Orlowski, T.E. Stinchcombe, B.S. Mitchell, T.C. Shea i inni. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. „J Clin Oncol”. 20 (22), s. 4420–4427, Nov 2002. PMID: 12431963.
- ↑ C. Aghajanian, S. Soignet, D.S. Dizon, C.S. Pien i inni. A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. „Clin Cancer Res”. 8 (8), s. 2505–2511, Aug 2002. PMID: 12171876.
- ↑ C.S. Chim, Y.Y. Hwang, C. Pang, T.W. Shek. Restoration of chemosensitivity by bortezomib: implications for refractory myeloma. „Nat Rev Clin Oncol”. 6 (4), s. 237–240, Apr 2009. DOI: 10.1038/nrclinonc.2009.15. PMID: 19333230.
- ↑ S. Jagannath, B. Barlogie, J. Berenson, D. Siegel i inni. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. „Br J Haematol”. 127 (2), s. 165–172, Oct 2004. DOI: 10.1111/j.1365-2141.2004.05188.x. PMID: 15461622.
- ↑ M.A. Dimopoulos, P.G. Richardson, R. Schlag, N.K. Khuageva i inni. VMP (Bortezomib, Melphalan, and Prednisone) is active and well tolerated in newly diagnosed patients with multiple myeloma with moderately impaired renal function, and results in reversal of renal impairment: cohort analysis of the phase III VISTA study. „J Clin Oncol”. 27 (36), s. 6086–6093, Dec 2009. DOI: 10.1200/JCO.2009.22.2232. PMID: 19858394.
- ↑ G. Avvisati. Bortezomib plus melphalan and prednisone for multiple myeloma. „N Engl J Med”. 359 (24), s. 2613; author reply 2613–4, Dec 2008. PMID: 19090032.
- ↑ R. Niesvizky, P.G. Richardson, S.V. Rajkumar, M. Coleman i inni. The relationship between quality of response and clinical benefit for patients treated on the bortezomib arm of the international, randomized, phase 3 APEX trial in relapsed multiple myeloma. „Br J Haematol”. 143 (1), s. 46–53, Sep 2008. DOI: 10.1111/j.1365-2141.2008.07303.x. PMID: 18673366.
- ↑ J. Koreth, K.E. Stevenson, H.T. Kim, M. Garcia i inni. Bortezomib, tacrolimus, and methotrexate for prophylaxis of graft-versus-host disease after reduced-intensity conditioning allogeneic stem cell transplantation from HLA-mismatched unrelated donors. „Blood”. 114 (18), s. 3956–3959, Oct 2009. DOI: 10.1182/blood-2009-07-231092. PMID: 19713456.
- ↑ S.P. Treon, L. Ioakimidis, J.D. Soumerai, C.J. Patterson i inni. Primary therapy of Waldenström macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. „J Clin Oncol”. 27 (23), s. 3830–3835, Aug 2009. DOI: 10.1200/JCO.2008.20.4677. PMID: 19506160.
- ↑ Daniel Lednicer, Lester A. Mitscher: The organic chemistry of drug synthesis. Volume 7. New York: Wiley, 2008, s. 133–134. ISBN 978-0-470-10750-8.
- ↑ P. Moreau, V. Coiteux, C. Hulin, X. Leleu i inni. Prospective comparison of subcutaneous versus intravenous administration of bortezomib in patients with multiple myeloma. „Haematologica”. 93 (12), s. 1908–1911, Dec 2008. DOI: 10.3324/haematol.13285. PMID: 18768528.
- ↑ C. Green, J. Bryant, A. Takeda, K. Cooper i inni. Bortezomib for the treatment of multiple myeloma patients. „Health Technol Assess”. 13 Suppl 1, s. 29–33, Jun 2009. DOI: 10.3310/hta13suppl1/05. PMID: 19567211.
- ↑ S. Lonial, E.K. Waller, P.G. Richardson, S. Jagannath i inni. Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. „Blood”. 106 (12), s. 3777–3784, Dec 2005. DOI: 10.1182/blood-2005-03-1173. PMID: 16099887.
- ↑ F. Lanzani, L. Mattavelli, B. Frigeni, F. Rossini i inni. Role of a pre-existing neuropathy on the course of bortezomib-induced peripheral neurotoxicity. „J Peripher Nerv Syst”. 13 (4), s. 267–274, Dec 2008. DOI: 10.1111/j.1529-8027.2008.00192.x. PMID: 19192066.
- ↑ K. Colson, D.S. Doss, R. Swift, J. Tariman i inni. Bortezomib, a newly approved proteasome inhibitor for the treatment of multiple myeloma: nursing implications. „Clin J Oncol Nurs”. 8 (5), s. 473–480, Oct 2004. PMID: 15515281.
- ↑ A. Hacihanefioglu, P. Tarkun, E. Gonullu. Acute severe cardiac failure in a myeloma patient due to proteasome inhibitor bortezomib. „Int J Hematol”. 88 (2), s. 219–222, Sep 2008. DOI: 10.1007/s12185-008-0139-7. PMID: 18633693.
- ↑ C. Cañete, A. Soley, M. Vuelta Arce, L. Escoda. Pulmonary toxicity after bortezomib. „Farm Hosp”. 32 (5). s. 301–302. PMID: 19150049.
- ↑ M.A. Dimopoulos, E. Kastritis, L. Rosinol, J. Bladé i inni. Pathogenesis and treatment of renal failure in multiple myeloma. „Leukemia”. 22 (8), s. 1485–1493, Aug 2008. DOI: 10.1038/leu.2008.131. PMID: 18528426.
- ↑ M. Furtado, S. Rule. Bortezomib-associated tumor lysis syndrome in multiple myeloma. „Leuk Lymphoma”. 49 (12), s. 2380–2382, Dec 2008. DOI: 10.1080/10428190802484099. PMID: 19052991.
- ↑ D.E. Reece, V. Sanchorawala, U. Hegenbart, G. Merlini i inni. Weekly and twice-weekly bortezomib in patients with systemic AL amyloidosis: results of a phase 1 dose-escalation study. „Blood”. 114 (8), s. 1489–1497, Aug 2009. DOI: 10.1182/blood-2009-02-203398. PMID: 19498019.
- ↑ D.I. Quinn, J. Nemunaitis, J. Fuloria, C.D. Britten i inni. Effect of the cytochrome P450 2C19 inhibitor omeprazole on the pharmacokinetics and safety profile of bortezomib in patients with advanced solid tumours, non-Hodgkin’s lymphoma or multiple myeloma. „Clin Pharmacokinet”. 48 (3), s. 199–209, 2009. DOI: 10.2165/00003088-200948030-00006. PMID: 19385713.
- ↑ T.Y. Kim, J. Park, B. Oh, H.J. Min i inni. Natural polyphenols antagonize the antimyeloma activity of proteasome inhibitor bortezomib by direct chemical interaction. „Br J Haematol”. 146 (3), s. 270–281, Aug 2009. DOI: 10.1111/j.1365-2141.2009.07752.x. PMID: 19500098.
- ↑ E.B. Golden, P.Y. Lam, A. Kardosh, K.J. Gaffney i inni. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. „Blood”. 113 (23), s. 5927–5937, Jun 2009. DOI: 10.1182/blood-2008-07-171389. PMID: 19190249.
- ↑ G. Perrone, T. Hideshima, H. Ikeda, Y. Okawa i inni. Ascorbic acid inhibits antitumor activity of bortezomib in vivo. „Leukemia”. 23 (9), s. 1679–1686, Sep 2009. DOI: 10.1038/leu.2009.83. PMID: 19369963.
- ↑ M. Basler, C. Lauer, U. Beck, M. Groettrup. The proteasome inhibitor bortezomib enhances the susceptibility to viral infection. „J Immunol”. 183 (10), s. 6145–6150, Nov 2009. DOI: 10.4049/jimmunol.0901596. PMID: 19841190.
- ↑ D. Schiff, P.Y. Wen, M.J. van den Bent. Neurological adverse effects caused by cytotoxic and targeted therapies. „Nat Rev Clin Oncol”. 6 (10), s. 596–603, Oct 2009. DOI: 10.1038/nrclinonc.2009.128. PMID: 19707193.
- ↑ L.C. Smith, P. Bertolotti, K. Curran, B. Jenkins. Gastrointestinal side effects associated with novel therapies in patients with multiple myeloma: consensus statement of the IMF Nurse Leadership Board. „Clin J Oncol Nurs”. 12 (3 Suppl), s. 37–52, Jun 2008. DOI: 10.1188/08.CJON.S1.37-51. PMID: 18490256.
- ↑ L. Pour, Z. Adam, L. Buresova, M. Krejci i inni. Varicella-zoster virus prophylaxis with low-dose acyclovir in patients with multiple myeloma treated with bortezomib. „Clin Lymphoma Myeloma”. 9 (2), s. 151–153, Apr 2009. DOI: 10.3816/CLM.2009.n.036. PMID: 19406726.
- ↑ A. Tanguy-Schmidt, M. Avenel-Audran, A. Croué, S. Lissandre i inni. [Bortezomib-induced acute neutrophilic dermatosis]. „Ann Dermatol Venereol”. 136 (5), s. 443–446, May 2009. DOI: 10.1016/j.annder.2008.11.021. PMID: 19442803.
- ↑ M.J. Wondergem, K. Grünberg, B.P. Wittgen, P. Sonneveld i inni. Interstitial pneumonitis caused by Pneumocystis jirovecii pneumonia (PCP) during bortezomib treatment. „Histopathology”. 54 (5), s. 631–633, Apr 2009. DOI: 10.1111/j.1365-2559.2009.03263.x. PMID: 19302537.
- ↑ Charakterystyka Produktu Leczniczego Velcade. [dostęp 2016-09-15].
- ↑ P.G. Richardson, P. Sonneveld, M.W. Schuster, E.A. Stadtmauer i inni. Reversibility of symptomatic peripheral neuropathy with bortezomib in the phase III APEX trial in relapsed multiple myeloma: impact of a dose-modification guideline. „Br J Haematol”. 144 (6), s. 895–903, Mar 2009. DOI: 10.1111/j.1365-2141.2008.07573.x. PMID: 19170677.
Bibliografia
- David E. Thurston: Chemistry and pharmacology of anticancer drugs. Boca Raton: CRC Press/Taylor & Francis, 2007, s. 121–122. ISBN 0-8493-9219-5.
- The chemotheraphy source book. Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins, 2008. ISBN 978-0-7817-7328-7.
- Kenneth Anderson, Irene M. Ghobrial: Multiple Myeloma: Translational and Emerging Therapies (Translational Medicine). Informa HealthCare. ISBN 978-1-4200-4510-9.
- Carmen Avendaño, J. Carlos Menendéz: Medicinal chemistry of anticancer drugs. Amsterdam: Elsevier, 2008. ISBN 978-0-444-52824-7.
- Indeks leków Medycyny Praktycznej 2009. Kraków: Wydawnictwo Medycyna Praktyczna, 2009. ISBN 978-83-7430-221-0. – Bortezomib, [w:] Indeks Leków MP, opis substancji, Medycyna Praktyczna [dostęp 2010-01-10].
- Charakterystyka Produktu Leczniczego Velcade. [dostęp 2016-09-15].
- Jan Kazimierz (-1997) Podlewski, Alicja Chwalibogowska-Podlewska, Robert Adamowicz: Leki współczesnej terapii. Warszawa: Split Trading, 2005, s. 87–88. ISBN 83-85632-82-4.
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.