Reakcja następcza – złożona reakcja chemiczna, w której produkty powstają z substratów dwuetapowo lub wieloetapowo, poprzez produkty przejściowe, powstające w kolejnych reakcjach elementarnych. W tych etapach reakcji następczej mogą zachodzić reakcje proste, odwracalne lub równoległe[1][2][3], np.:
- A + B → C + D,
- C ⇌ E + F,
- C + D ⇌ G.
Równanie reakcji następczej (jak każdej reakcji złożonej) jest sumą odpowiednich równań reakcji elementarnych – wyraża bilans masy (zobacz – stechiometria), a nie ilustruje mechanizmu reakcji. Wyrażenie określające wartość stałej równowagi reakcji następczej jest liniową kombinacją wyrażeń dotyczących reakcji elementarnych[4]. Jeżeli różnica między szybkościami obu reakcji elementarnych jest duża, szybkość reakcji złożonej jest równa szybkości jej najwolniejszego etapu[2].
Elementy kinetyki reakcji następczych
Sposób analizy szybkości reakcji następczych zostanie przedstawiony na przykładzie procesu dwuetapowego, złożonego z dwóch reakcji prostych pierwszego rzędu[2][3]:
- A → B → C.
Jeżeli proces zachodzi w układzie termodynamicznie zamkniętym sumaryczne stężenie wszystkich trzech reagentów nie ulega zmianom w czasie, a więc w każdej chwili jest spełniona zależność[2][3]:

- a = cA + cB + cC,
gdzie – początkowe stężenie substratu A.

Szybkość reakcji wyraża się np. jako szybkość zmian stężenia substratu lub produktu w czasie. W przypadku reakcji I rzędu szybkość zmian stężenia substratu (A) w wyniku reakcji pierwszej (A → B, stała szybkości reakcji – ) wyraża równanie[2][3]:


lub (po scałkowaniu):

lub (po przekształceniu):

Wykresem funkcji jest krzywa logarytmiczna w układzie współrzędnych lub prosta w układzie (o współczynniku kierunkowym ).


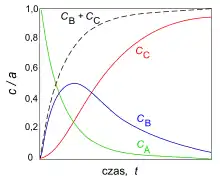
Produkt pośredni B (produkt reakcji 1 i substrat reakcji 2) początkowo gromadzi się w układzie, ponieważ szybkość reakcji A → B jest większa od B → C. W tym okresie zmniejsza się stężenie a stężenie rośnie, co powoduje zmniejszanie się szybkości reakcji 1 i wzrost szybkości reakcji 2 (powstawanie produktu końcowego C). W kolejnym okresie stężenie zbliża się do zera, a osiąga wartość maksymalną w danych warunkach zewnętrznych. W następnym okresie obserwuje się stopniowe zmniejszanie się stężenia i wzrost stężenia przy czym[2][3]:






Kształt krzywej zależy od stosunku stałych szybkości obu reakcji elementarnych ( i ), co wyraża zależność[2]:




Elementy termodynamiki chemicznej
W większości układów rozpatruje się możliwości równoczesnego przebiegu elementarnych reakcji chemicznych w obu kierunkach (z różnymi szybkościami, łącznie – w kierunku stanu równowagi). Ilustruje to przykład procesu[4]:
- A + B ⇌ C + D,
- C + B ⇌ D + E.
Jako przykład takiej reakcji jest podawany złożony proces bezciśnieniowej konwersji metanu parą wodną na katalizatorze niklowym w temperaturze 600–800 °C. W tym przypadku, istnieje wiele możliwych do pomyślenia reakcji elementarnych, lecz w warunkach procesu (m.in. temperatura, nadmiar wody) istotną rolę odgrywają dwie reakcje odwracalne, biegnące szybciej w prawo[uwaga 1][4]:
- CH4 + H2O ⇌ CO + 3 H2,
- CO + H2O ⇌ CO2 + H2.
Stężenia poszczególnych reagentów w stanie równowagi są obliczane z wykorzystaniem informacji stałych równowagi obu etapów oraz wartości stopnia przemiany dwutlenku węgla i metanu (względny stopień przereagowania, związany z liczbą postępu reakcji)[4].
Zobacz też
Uwagi
- ↑ Termodynamika chemiczna pozwala ustalać, które z wielu reakcji prostych, które mogą jednocześnie przebiegać w mieszaninie reagentów, decydują o przebiegu reakcji złożonej. Spośród wszystkich równań reakcji, które są możliwe do pomyślenia, w pierwszej kolejności odrzuca się te, które można przedstawić jako liniowe kombinacje innych. Dla pozostałych reakcji oblicza się – na podstawie danych termodynamicznych (zobacz: Równanie van ’t Hoffa) – wartości stałych równowagi w danych warunkach zewnętrznych, co pozwala odrzucić te reakcje, dla których wartości są najmniejsze (ich produkty mogą występować tylko w ilościach śladowych, np. mniejszych od granicy wykrywalności). W kolejnym etapie selekcji uwzględnia się informacje o stężeniach reagentów i określa kierunek samorzutnego przebiegu każdej z reakcji [Józef Szarawara, s. 403–405].


Przypisy
- ↑ Leksykon naukowo-techniczny z suplementem. T. P–Ż. Warszawa: WNT, 1989, s. 806. ISBN 83-204-0969-1.
- 1 2 3 4 5 6 7 Stanisław Bursa: Chemia fizyczna. Wyd. Wyd. 2 popr. Warszawa: Państwowe Wydawnictwo Naukowe, 1979, s. 589–670. ISBN 83-01-00152-6.
- 1 2 3 4 5 Antoni Basiński i wsp.: Chemia fizyczna. Warszawa: Państwowe Wydawnictwo Naukowe, 1966, s. 574–576.
- 1 2 3 4 Józef Szarawara: Termodynamika chemiczna. Warszawa: WNT, 1969, s. 383–385.