

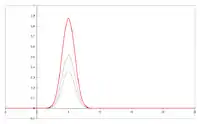
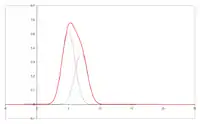
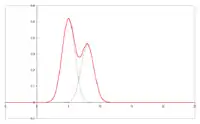

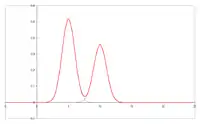



Kolor:
zielony – wyraz dyfuzji wirowej

czerwony – wyraz dyfuzji podłużnej

niebieski – wyraz oporów przenikania masy

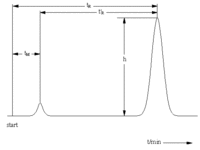
tR – czas retencji związków nie zatrzymywanych w kolumnie
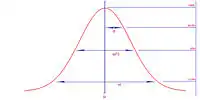
(sposób określania W1/2 i odchylenia standardowego σ)
Równanie van Deemtera – funkcja określająca zależność wielkości półki teoretycznej (WRPT lub HETP, height equivalent to a theoretical plate) w kolumnie GC od liniowej prędkości gazu nośnego i kinetyki wymiany masy między gazem nośnym i fazą nieruchomą (filmem nielotnej cieczy na stałym nośniku), w tym od współczynników dyfuzji w obu fazach.
Liczba półek teoretycznych kolumny chromatograficznej jest miarą sprawności rozdzielania analizowanych mieszanin na pojedyncze składniki w określonych warunkach[1].
Zasada chromatografii gazowej
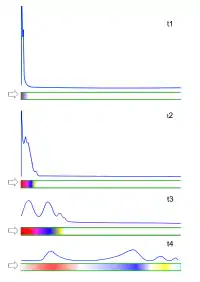
Chromatografia gazowa jest metodą analizy mieszanin związków chemicznych, które są gazami w temperaturze pracy kolumny. Analizowaną próbkę wprowadza się, w jak najkrótszym czasie, do kolumny chromatograficznej, która zawiera sorbent stały lub trudnolotną ciecz, w postaci filmu pokrywającego ścianki kolumn kapilarnych lub ziarna nośnika. Składniki analizowanych mieszanin (anality) przenosi przez kolumnę strumień gazu nośnego, np. helu, argonu lub wodoru. Cząsteczki analitów przemieszczają się przez kolumnę z różną prędkością, która jest zależna od sił ich oddziaływań z fazą stacjonarną, np. od rozpuszczalności w cieczy, pokrywającej wewnętrzne ścianki kolumny kapilarnej. W przypadku stosowania niepolarnych faz stacjonarnych prędkość przemieszczania się cząsteczek analizowanego związku jest tym większa, im niższa jest jego temperatura wrzenia. Proces rozdzielania analitów może być traktowany jak szereg następujących kolejno stanów równowagi procesów sorpcji badanych związków i ich desorpcji do strumienia gazu nośnego (stopnie równowagi, półki teoretyczne).
Rozdzielone składniki analizowanej mieszaniny kolejno wypływają z kolumny do detektora, który reaguje na obecność cząsteczek innych niż cząsteczki gazu nośnego. Podstawą analiz ilościowych (oznaczeń składu) jest wielkość sygnału detektora, a analiz jakościowych – charakterystyczny dla określonego związku czas wymywania z kolumny (czas retencji) lub odpowiednia objętość gazu nośnego, potrzebna do wymycia związku (objętość retencji)[2][3].
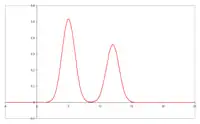
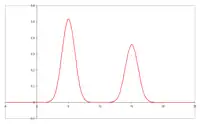
Szerokość i kształt piku GC
W czasie przemieszczania się próbki przez kolumnę zachodzi nie tylko wzajemne rozsuwanie się pasm poszczególnych związków. Zwiększa się też szerokość odpowiednich pasm i kształt rejestrowanych pików (tzw. „ogonowanie”). Kształt rejestrowanych pików chromatogramu jest podstawą wniosków, dotyczących właściwości powierzchni sorbentów (metoda chromatografii inwersyjnej).
Rozmycie pasm wpływa niekorzystnie na możliwości rozdzielenia składników w celach analitycznych, czyli sprawność kolumny. W przypadku chromatografii ciecz–gaz głównymi przyczynami rozszerzania się pasm są:
- dyfuzja cząsteczek analitów wzdłuż kolumny – w obu kierunkach (dyfuzja podłużna),
- dyfuzja wirowa gazu nośnego,
- opory przenoszenia masy w fazie stacjonarnej, zależne np. od grubości filmu cieczy i współczynników dyfuzji analitów.
Równanie van Deemtera
Równanie wyraża wpływy różnych czynników na szerokość piku i wysokość równoważną półce teoretycznej (WRPT). Zostało po raz pierwszy opublikowane przez van Deemtera, Zuiderwega i Klinkenberga w 1956[4]. Jest znane w formie:

gdzie:
- – liniowa prędkość przepływu gazu nośnego (np. m/s), mierzona na wylocie z kolumny,
- – wyraz dyfuzji wirowej,
- – wyraz dyfuzji podłużnej,
- – wyraz oporów przenikania masy między fazą stacjonarną i ruchomą.



Funkcja ma minimum w punkcie:

Stosowanie w równaniu jednej wartości liniowej prędkości przepływu gazu nośnego jest uproszczeniem. W rzeczywistości prędkość nie jest stała. Gaz przepływa wzdłuż kolumny dzięki dość dużej różnicy ciśnień, zależnej od rodzaju i długości kolumny. Początkowa wartość ciśnienia stopniowo zmniejsza się, wskutek pokonywania oporów przepływu (zgodnie z równaniem Darcy’ego-Weisbacha), co wiąże się ze zmianą objętości gazu i liniowej prędkości przepływu. Efekt ten jest uwzględniony w zmodyfikowanych wersjach równania (np. J.C. Giddings 1963)[3].
Wyraz A jest prawie niezależny od prędkości przepływu gazu nośnego. Jest wiązany ze zróżnicowaniem długości różnych linii prądu (strug).
Wartość A zależy w dużym stopniu od rodzaju fazy stacjonarnej (porowatość, wymiary i kształt ziaren, sposób ich upakowania w kolumnie) oraz od stosunku średnicy ziarna do średnicy kolumny. W przypadku kolumn kapilarnych, w których fazą stacjonarną jest film cieczy, pokrywający wewnętrzne ścianki, czynnik dyfuzji wirowej jest równy zeru.
Wyraz B jest wiązany z przypadkowymi ruchami cząsteczek analitów w strumieniu gazu nośnego. Wpływ tych efektów na stopień rozmycia piku jest tym mniejszy, im większa jest prędkość gazu nośnego i krótszy czas przebywania składników analizowanej próbki w kolumnie (linia B jest malejąca). Stopnień rozmycia pasm wskutek dyfuzji podłużnej zależy od gęstości gazu nośnego. Z tego punktu widzenia korzystne jest stosowanie wodoru i helu, a nie azotu i argonu, nie zawsze jest to jednak wybór optymalny. Konieczne jest uwzględnienie wpływu rodzaju gazu nośnego na czas retencji i czułość detektorów.
Wyraz C ma związek z istnieniem oporów przenikania cząsteczek analitu przez granicę między fazą ruchomą i nieruchomą. Proces przenikania obejmuje etapy dyfuzji w fazie gazowej do granicy faz, przenikania przez tę granicę i dyfuzji w fazie stacjonarnej. W każdym z układów faza stacjonarna–faza ruchoma kinetyka poszczególnych etapów może mieć inny wpływ na łączną wartość wyrazu C.
Wyznaczanie wysokości półki teoretycznej w praktyce
W praktyce wysokość półki teoretycznej oblicza się na podstawie wyników pomiarów liczby tych półek (N) w badanej kolumnie:

Liczbę półek teoretycznych wyznacza się na podstawie:
- czasu retencji i stopnia rozmycia pasma (odchylenie standardowe dla krzywej Gausse’a, za jaką uznaje się pik)



- szerokości piku GC w połowie wysokości lub u podstawy




Przypisy
- ↑ Compendium of Chemical Terminology, IUPAC Gold Book, wyd. 2, www.iupac.org, 1997 [dostęp 2010-12-31] [zarchiwizowane z adresu 2009-09-19] (ang.).
- ↑ Uniwersytet Gdański, Wydział Chemii, Katedra Analizy Środowiska: Chromatografia gazowa. 2007. [dostęp 2010-12-31]. (pol.).
- 1 2 Orion Edwin Schupp III (tłum. Jerzy Kuryłowicz): Chromatografia gazowa. Warszawa: Państwowe Wydawnictwo Naukowe, 1972, s. 72–113.
- ↑ van Deemter JJ, Zuiderweg FJ and Klinkenberg A. Longitudinal diffusion and resistance to mass transfer as causes of non ideality in chromatography. „Chem. Eng. Sci.”. 5, s. 271–289, 1956. DOI: 10.1016/0009-2509(56)80003-1.
Bibliografia
- Zygfryd Witkiewicz, Jacek Hepter: Chromatografia gazowa. Wyd. 2. Warszawa: Wydawnictwa Naukowo-Techniczne, 2009. (pol.).