Oczyszczanie białek – szereg procesów rozdziału mający na celu izolację pojedynczego typu białka ze złożonej mieszaniny[1].
Izolując dane białko z komórki, należy je odseparować od tysięcy innych białek, które się w niej znajdują. Metody oddzielania jednych cząsteczek od drugich opierają się różnicy w jednej lub wielu fizycznych bądź chemicznych właściwościach. Im więcej jest tych różnic i im są większe, tym łatwiej przebiega proces izolacji. W przypadku oczyszczania białek najczęściej wykorzystuje się różnice w wielkości (długości łańcucha polipeptydowego lub masy), powinowactwie do specyficznych ligandów[2], a także różnice w ładunku i hydrofobowości.
Postępowanie przed oczyszczaniem
Materiałem wyjściowym do izolacji białka powinien być materiał biologiczny o jak największej zawartości pożądanego białka i możliwie ubogi w zanieczyszczenia. Białka przeznaczone do oczyszczania uwalnia się do roztworu[3]. Jeśli białka wydzielane są pozakomórkowo, należy usunąć biomasę poprzez np. filtrację, wirowanie, flotację[4]. Jeśli są to białka wewnątrzkomórkowe, np. występujące w organellach komórkowych bądź związane z błonami komórkowymi, wymagane jest rozbicie tkanek i doprowadzenie do dezintegracji komórek, aby rozpuścić białka w roztworze. W tym celu stosuje się m.in. homogenizację, sonikację (ultradźwięki), trawienie enzymatyczne, lizę osmotyczną, ucieranie z materiałami ściernymi (np. piaskiem), działanie zasad, detergentów, rozpuszczalników organicznych, wysokich ciśnień (prasa Frencha)[3].
Optymalizacja oczyszczania białek
Aby uzyskać wyższy stopień czystości białek zazwyczaj stosuje się kombinację różnych technik opartych na różnicy w ich właściwościach[5]. Wymagany stopień czystości białka zależy od jego przeznaczenia. Przykładowo dla zastosowań terapeutycznych wymagana jest bardzo wysoka czystość (>99%), w celu przeprowadzenia krystalografii i dla większości badań fizykochemicznych – wysoka (95–99%), a przy wykorzystaniu do produkcji przeciwciał – umiarkowana (<95%)[3][6].
Najważniejszymi parametrami w oczyszczaniu białek są:
- rozdzielczość – zdolność do oddzielania od siebie cząsteczek; powinna być wysoka w ostatnim etapie oczyszczania, kiedy zanieczyszczenia i pożądane białko wykazują wiele podobnych właściwości;
- pojemność – określa objętość próbki, jaka może być oczyszczana (np. załadowana do kolumny) bez utraty jakości procesu (może być ograniczana np. przez wysoką zawartość zanieczyszczeń w próbce);
- szybkość – ma największe znaczenie na początku procesu, kiedy niektórych zanieczyszczeń (np. proteaz) należy się pozbyć jak najszybciej;
- odzysk białka – może się obniżać np. przez nieodpowiednie warunki, procesy degradacyjne. Każdy dodatkowy etap oczyszczania zwiększa czystość, ale zmniejsza ilość odzyskanego białka[7].
Optymalizowanie każdego z tych czterech parametrów może odbyć się tylko kosztem innego, dlatego przy oczyszczaniu należy znaleźć kompromis[7].
Czasami zadowalający poziom czystości można osiągnąć w jednoetapowym oczyszczaniu; w innych przypadkach można zastosować 3-etapową strategię oczyszczania CIPP (ang. Capture, Intermediate Purification, and Polishing). Pozwala ona zaplanować, w jaki sposób połączyć różne metody oczyszczania, aby osiągnąć najlepszy efekt[7]. Etapy oczyszczania białka są następujące:
- przygotowanie roztworu białka do oczyszczenia, które może obejmować ekstrakcję, klaryfikację;
- oczyszczanie właściwe (ang. capture) – izolowanie, zatężanie i stabilizowanie pożądanego białka; zwykle optymalizowana jest na tym etapie szybkość i pojemność;
- etap pośredniego oczyszczania (ang. intermediate purification) – usuwanie większości zanieczyszczeń takich jak inne białka, kwasy nukleinowe, endotoksyny, wirusy; jeśli oczyszczanie właściwe przebiegło efektywnie, można ten etap pominąć. Najważniejszymi parametrami na tym etapie są rozdzielczość i pojemność;
- doczyszczanie (ang. polishing) – usuwanie pozostałych śladowych ilości zanieczyszczeń, agregatów białkowych, białek o podobnych właściwościach. Najważniejszym parametrem tego etapu jest rozdzielczość[7][8][9].
Każdy kolejny etap powoduje jednak utratę białek i może niekorzystnie wpływać na ich aktywność. Znajomość parametrów krytycznych dla stabiności białka (m.in. temperatura, pH, ciśnienie, siła jonowa, obecność kofaktorów) pomaga w minimalizowaniu strat[3]. Generalnie lepiej jest otrzymać białko nadmiernie oczyszczone niż niedostatecznie oczyszczone, ponieważ niski stopień czystości, nieznane zanieczyszczenia mogą fałszować wyniki[9]. Największy problem przy oczyszczaniu białka stanowi proteoliza. Można ją ograniczyć stosując inhibitory proteaz i obniżoną temperaturę[10].
Techniki rozdzielania białek
Rozdział ze względu na wielkość
Wirowanie
Użycie wirówek pozwala rozdzielić cząstki w roztworze (np. organelle komórkowe, cząsteczki) różniące się masą lub gęstościami. Osiadają one bowiem na dno probówki w różnym tempie – cięższe lub o większej gęstości osiadają szybciej. Białka mogą bardzo różnić się między sobą masą, ale nie gęstością (jeśli nie są połączone z lipidami czy węglowodanami, zwykle ich gęstość mieści się w granicach 1,37 g/cm3 ± 15%). Ultrawirówki mogące uzyskać przyspieszenie 1 000 000 × g potrafią efektywnie rozdzielać cząstki o rozmiarach do 10 kDa; cząstki o mniejszych rozmiarach nie będą osadzać się równomiernie[2].
Wirowanie frakcjonujące powszechnie stosuje się na początkowym etapie oczyszczania białek, aby oddzielić rozpuszczalne białka od nierozpuszczalnych części komórki. Po odwirowaniu komórkowego homogenatu otrzymuje się osad na dnie probówki złożony z organelli komórkowych oraz roztwór białek w supernatancie[2].
Białka różniące się od siebie masami mogą być oddzielone przez wirowanie w gradiencie gęstości, do wytworzenia którego zwykle wykorzystuje się roztwór sacharozy. Białka naniesione na powierzchnię roztworu o zmieniającym się stężeniu sacharozy migrują w kierunku dna probówki zgodnie ze swoimi współczynnikami sedymentacji. Ostatecznie tworzą w probówce prążki lub strefy zależne od ich masy. Jeśli wirowanie trwa zbyt krótko, cząstki nie przemieszczą się wystarczająco, by się dobrze od siebie oddzielić; jeśli zbyt długo – wszystkie cząstki znajdą się na dnie[2].
Filtracja żelowa
Filtracja żelowa (chromatografia żelowa, sączenie molekularne) polega na rozdziale białek w kolumnach wypełnionych porowatym polimerem jak np. poliakrylamid (Biogel), dekstran (Sephadex), agaroza (Sepharose), tj. przez tzw. sita molekularne[3][11]. Małe cząsteczki dyfundują w głąb żelu i wolniej migrują w kolumnie. Większe cząsteczki takie jak białka, o większym rozmiarze od porów ziaren wypełniacza nie wnikają do nich i znajdują się tylko w roztworze wodnym wypełniającym przestrzenie między ziarnami, przez co szybciej wypływają z kolumny[5]. W przeciwieństwie do innych chromatograficznych metod rozdziału cząstki nie wiążą się z wypełniaczem kolumny i skład buforu nie ma wpływu na rozdział[3].
Rozdział ze względu na ładunek
Suma dodatnich i ujemnych ładunków bocznych reszt aminokwasowych tworzy ładunek wypadkowy cząsteczki białka. Ładunek ten można zobojętnić, dlatego każde białko ma punkt izoelektryczny (pI), czyli takie pH, w którym wypadkowy ładunek białka jest równy zero. W takich warunkach rozpuszczalność białek jest najmniejsza[5].
Elektroforeza
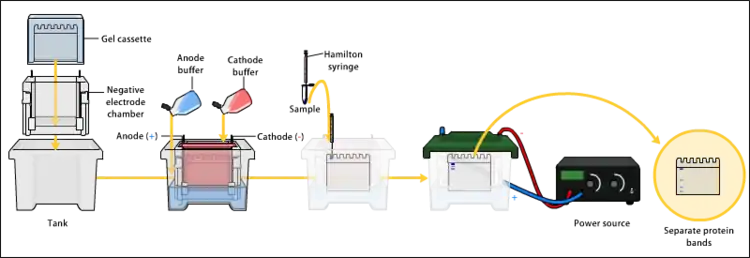
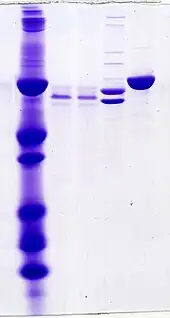
Elektroforeza jest techniką używaną do oddzielania cząsteczek pod wpływem pola elektrycznego. Cząsteczki migrują w tempie zależnym od stosunku ładunek:masa. Przykładowo przy takiej samej masie i kształcie, szybciej w stronę elektrody będą przemieszczać się cząsteczki o wyższym ładunku wypadkowym[2]. Na tej zasadzie białka nałożone na nośnik (żel) migrują w polu elektrycznym w różnym tempie w kierunku anody lub katody. Po rozdziale można je wyekstrahować z żelu[5].
Zazwyczaj jako nośnika używa się żelu poliakrylamidowego. Wielkość porów żelu można dostosowywać, modyfikując stężenie poliakrylamidu i czynnika sieciującego, co wraz z siłą pola elektrycznego ma wpływ na prędkość przemieszczania się białek. Przed lub podczas elektroforezy można wystawić białka na działanie detergentu SDS, który je denaturuje, niszczy czwartorzędową strukturę białka (powoduje dysocjację tworzących je łańcuchów polipeptydowych)[2], a także trzeciorzędową strukturę białka (sprawia, że nabierają struktury liniowej) i nadaje ujemny ładunek[12]. Jedynym czynnikiem wpływającym na tempo migracji białek w elektroforezie w żelu poliakrylamidowym w obecności SDS jest długość łańcucha (co jest powiązane z jego masą)[2].
O ile za pomocą elektroforezy z zastosowaniem SDS można rozdzielić białka różniące się masą, tak w przypadku podobnych mas można wykorzystać różnice w wypadkowym ładunku elektrycznym. Pozwala na to dwuwymiarowa elektroforeza żelowa, w której białka najpierw rozdzielane są ze względu na ładunek, a w drugim etapie ze względu na masę[2]. Zdenaturowane przez stężony mocznik białka przemieszczają się w żelu o gradiencie pH uzyskanym przez zastosowanie amfolitów i zatrzymują się w miejscu, gdzie pH równe jest ich punktowi izoelektrycznemu. Wtedy ich wypadkowy ładunek wynosi zero i nie mogą się dalej przemieszczać w polu elektrycznym. Technika ta nazywana jest ogniskowaniem izoelektrycznym[5][2]. Tak rozdzielone białka mogą być dalej rozdzielane ze względu na swoją masę. Żel ten można umieścić na żelu poliakrylamidowym i przeprowadzić elektroforezę w obecności SDS[2].
Elektroforeza jest używana głównie w celach analitycznych ze względu na trudności w rozdzielaniu w ten sposób dużych ilości białek i koszt urządzenia, który by to umożliwiał. Poza tym rozdział ze względu na ładunek czy punkt izoelektryczny umożliwia w prostszy sposób chromatografia jonowymienna[13].
Chromatografia jonowymienna
W chromatografii jonowymiennej kolumny wypełnione są nośnikiem o ładunku ujemnym lub dodatnim. Białka o takim samym ładunku wypadkowym jak nośnik odpychają się od niego i swobodnie przez niego przepływają. Zatem w przypadku nośnika naładowanego dodatnio, białka elektrycznie obojętne i naładowane dodatnio (zasadowe) przepłyną swobodnie przez kolumnę, a białka naładowane ujemnie (kwaśne) odwracalnie się z nim zwiążą. Związane białka są następnie wymywane przez przepływ roztworów o wzrastającej sile jonowej (gradient solny tworzony zwykle przez NaCl lub KCl). Przy niskim stężeniu soli białka i nośnik przyciągają się przez swoje różnoimienne ładunki. W wyższych stężeniach soli jej ujemne jony wiążą się do dodatnio naładowanego nośnika, wypierając ujemnie naładowane białka. Dzięki gradientowi (wzrastającemu stężeniu soli) słabo naładowane białka są wymywane w pierwszej kolejności, a białka o wysokim ładunku – jako ostatnie. Analogicznie nośnik naładowany ujemnie może służyć do rozdzielenia zasadowych białek[2][5].
Rozdział ze względu na hydrofobowość


Wysalanie białek
Generalnie białka posiadające dodatnio i ujemnie naładowane regiony mają tendencję do agregacji przy bardzo niskich stężeniach soli w środowisku. Kiedy umiarkowane ilości soli są dostępne, aniony i kationy neutralizują ładunki na powierzchni białek, co zapobiega tworzeniu się skupisk. Zjawisko to nazywa się wsalaniem i jest powiązane ze wzrostem rozpuszczalności białka pod wpływem dodatku soli. Kiedy stężenie soli jest wysokie, powierzchnia białek znów staje się naładowana[14], sól będzie odciągać wodę od białek, doprowadzając do hydratacji swoich jonów, przez co silniejsze staną się oddziaływania hydrofobowe białek. Cząsteczki białek w takich warunkach będą miały tendencję do tworzenia agregatów i wytrącania się z roztworu, co nazywane jest wysalaniem[15].
Białka z większymi obszarami hydrofobowymi będą potrzebować mniejszego stężenia soli, aby się wytrącać i różnicę tę można wykorzystać w oczyszczaniu białek. Najczęściej jako sól wykorzystuje się siarczan amonu, który pozwala zachować natywną strukturę białka[3]. Precypitację przeprowadza się w kilku etapach. Najpierw dodaje się siarczanu amonu w ilości niewystarczającej do wytrącenia pożądanego białka, ale zdolnej do wytrącenia innych niepożądanych białek. Po odwirowaniu pożądane białko będzie znajdować się w supernatancie. Następnie można dodać siarczanu amonu w ilości wystarczającej do wytrącenia pożądanego białka. Po wirowaniu będzie się ono znajdować w osadzie, który można rozpuścić w buforze, a supernatant usunąć[16]. Innym, lepszym sposobem na pozbycie się siarczanu po wysalaniu jest dializa[14]. Liczba etapów i ilość zastosowanego siarczanu amonu na każdym z nich trzeba wyznaczyć empirycznie[16].
Chromatografia oddziaływań hydrofobowych
Technika chromatografii oddziaływań hydrofobowych pozwala na rozdział białek różniących się siłą oddziaływania ich obszarów hydrofobowych z jeszcze bardziej hydrofobowymi grupami ligandów na powierzchni nośnika pozbawionego ładunku elektrycznego, w roztworach o dużej sile jonowej (np. 1,5 M siarczan amonu). Białka są uwalniane poprzez wymywanie buforem o zmniejszającym się gradiencie soli. Alternatywnie można je również eluować buforem zwiększającym hydrofilowość białek (np. alkohole, detergenty niejonowe) czy zawierającym substancje o silnym powinowactwie do ligandu[3].
Rozdział ze względu na powinowactwo

W rozdzielaniu białek można wykorzystać wysoką specyficzność oddziaływań receptor-ligand, przeciwciało-antygen, enzym-substrat[5]. Metodą wykorzystującą te właściwości jest chromatografia powinowactwa. Cząsteczki ligandu (który może być np. enzymem, przeciwciałem) związane kowalencyjnie z wypełnieniem kolumny wiążą specyficznie pożądane białko, a inne białka i zanieczyszczenia przepływają swobodnie przez kolumnę niezależnie od ich ładunku czy masy. Pożądane białka są następnie wymywane przez dodatek nadmiaru ligandu bądź przez zmianę stężenia soli lub pH[2].
Wykrywanie pożądanego białka po oczyszczaniu
W celu wykrycia obecności pożądanego białka w próbce wykorzystuje się jego charakterystyczne właściwości – zdolność do wiązania określonego liganda, katalizowania określonej reakcji czy bycia rozpoznanym przez specyficzne przeciwciało[17]. Do metod wykrywania białek należy m.in.:
- western blot – obejmuje elektroforetyczny rozdział białek, transport z żelu na membranę, inkubację z przeciwciałem pierwszorzędowym rozpoznającym pożądane białko, inkubację z przeciwciałem drugorzędowym skoniugowanym z enzymem, który po dodaniu substratu umożliwia wizualizację białka[2];
- zymografia – obejmuje elektroforetyczny rozdział białek i wizualną identyfikację enzymatycznej aktywności pożądanego białka. Pożądane białko musi być enzymem;
- spektroskopia masowa[17].
Techniki rekombinacyjne
Zwiększając ilość pożądanego białka w materiale biologicznym, zwiększa się efektywność jego oczyszczania. Można to zrobić, stosując nadekspresję zrekombinowanego genu białka w komórce. Dodatkowo można dodać odpowiednią sekwencję nukleotydową, aby zrekombinowane białko zawierało znacznik ułatwiający jego późniejsze oczyszczanie. Takim znacznikiem może być sześć histydyn (His-Tag), dzięki którym łatwo jest oczyścić białko metodą chromatografii metalopowinowactwa – nośnik na kolumnie ma unieruchomione jony metali, z którymi wiąże się ten fragment histydynowy[5].
Przypisy
- ↑ S. Shanmugam, T. Sathishkumar: Enzyme Technology. New Delhi: I. K International Publishing House Pvt. Ltd., 2009, s. 28. ISBN 978-93-80026-05-3.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 H. Lodish, A. Berk, P. Matsudaira, C. A. Kaiser, M. Krieger, M. P. Scott, L. Zipursky, J. Darnell: Molecular Cell Biology. W. H. Freeman, 2003, s. 86–92. ISBN 978-0-7167-4366-8.
- 1 2 3 4 5 6 7 8 M. Rosiński, D. Piasecka-Kwiatkowska, J. R. Warchalewski. Przegląd metod separacji i oczyszczania białek przydatnych w badaniach i analizie żywności. „Żywność. Nauka. Technologia. Jakość”. 3 (44), s. 5–22, 2005.
- ↑ B. K. Nfor, T. Ahamed, G. W. K. van Dedem, L. A. M. van der Wielen, E. J. A. X. van de Sandt, M. H. M. Eppink, M. Ottens. Design strategies for integrated protein purification processes: challenges, progress and outlook. „Journal of Chemical Technology and Biotechnology”. 83 (2), s. 124–132, 2008. DOI: 10.1002/jctb.1815.
- 1 2 3 4 5 6 7 8 P. C. Turner, A. G. McLennan, A. D. Bates, M. R. H. White: Krótkie wykłady. Biologia Molekularna. Warszawa: Wydawnictwo Naukowe PWN, 2009, s. 31–33. ISBN 978-83-01-14146-2.
- ↑ Protein Purification Strategies. GE Healthcare Life Sciences. [dostęp 2017-02-24]. [zarchiwizowane z tego adresu (2017-01-23)]. (ang.).
- 1 2 3 4 Strategies For Protein Purification. GE Healthcare Life Sciences. [dostęp 2017-02-24]. [zarchiwizowane z tego adresu (2017-03-18)]. (ang.).
- ↑ M. Olszewski, P. Filipkowski. Benzonaza® — możliwości praktycznych zastosowań. „Postępy Biochemii”. 55 (1), s. 21, 2009.
- 1 2 Protein Purification. Amersham Pharmacia Biotech, 1999. [dostęp 2017-02-24]. [zarchiwizowane z tego adresu (2016-11-30)]. (ang.).
- ↑ C. Ritchie. Protease Inhibitors. „Materials and Methods”, s. 3–169, 2013. DOI: 10.13070/mm.en.3.169.
- ↑ Sączenie Molekularne. stareaneksy.pwn.pl. [dostęp 2017-02-24]. (pol.).
- ↑ N. Oswald: How SDS-PAGE works. BitesizeBio, 2016. [dostęp 2017-02-24]. (ang.).
- ↑ R. K. Scopes: Protein Purification. Principles and Practice. New York: Springer, 1994, s. 250. DOI: 10.1007/978-1-4757-2333-5. ISBN 978-1-4419-2833-7.
- 1 2 K. C. Duong-Ly, S. B. Gabelli. Chapter Seven – Salting out of Proteins Using Ammonium Sulfate Precipitation. „Methods in Enzymology”. 541, s. 85–94, 2014. DOI: 10.1016/B978-0-12-420119-4.00007-0.
- ↑ Protein Fractionation Techniques. Stony Brook University, 1999. [dostęp 2017-02-24]. [zarchiwizowane z tego adresu (2016-12-19)]. (ang.).
- 1 2 Ammonium Sulfate Solution, 4.1 M Product Information. Sigma. [dostęp 2017-02-24]. (ang.).
- 1 2 R. K. Scopes: Protein Purification. Principles and Practice. New York: Springer, 1994, s. 303–309. DOI: 10.1007/978-1-4757-2333-5. ISBN 978-1-4419-2833-7.