| Klasyfikacje | |
| ICD-10 | |
|---|---|
Malformacje odbytu i odbytnicy (ang. anorectal malformations) – wady wrodzone dystalnego (dalszego) odcinka przewodu pokarmowego: odbytu i odbytnicy, a także układu moczowo-płciowego u obu płci. Częstość tych wad szacuje się na 1:5000 żywych urodzeń. Wady te są różne: od drobnych, które leczone mają doskonałe rokowanie, do ciężkich, złożonych malformacji trudnych do leczenia, nierzadko skojarzonych z innymi wadami obarczonymi fatalnym rokowaniem. Począwszy od początku lat 80., gdy wprowadzono nowe techniki operacyjne, bardzo poprawiły się rokowania u dzieci z tego typu wadami; chirurdzy mogli łatwiej ocenić stosunki anatomiczne przewodu pokarmowego i dróg moczowo-płciowych. Poprawę przeżywalności dzieci z tymi wadami przypisuje się także postępowi w technikach obrazowania i w wiedzy o budowie i fizjologii struktur anatomicznych miednicy noworodka.
Historia

Zarośnięty odbyt był dobrze znaną wadą wrodzoną od stuleci, i przez stulecia stosowano prymitywną metodę leczenia wady polegającą na utworzeniu otworu w kroczu dziecka. W Polsce jeden z najwcześniejszych opisów wrodzonego zarośnięcia odbytu znalazł się w dziele Jana Chrościejewskiego, wykładowcy Akademii Weneckiej, „De morbis puerorum tractatem locupletissimi” (1583).
Dzieci z wadą która dziś określona byłaby jako „niska” przeżywały, te które miały wadę „wysoką” – nie[2]. Francuski chirurg Jean Zuléma Amussat w 1835 roku jako pierwszy przeprowadził operację zespolenia ściany odbytnicy i brzegów otworu w skórze krocza[3], a operacja zwana później jako operacja Amussata, była pierwowzorem przeprowadzanej dziś anoplastyki.
Przez pierwsze sześćdziesiąt lat XX wieku chirurdzy wykonywali operację krocza bez kolostomii dla „niskich” wad. „Wysokie” malformacje leczono z wytworzeniem kolostomii w okresie noworodkowym, i późniejszym połączeniem brzuszno-kroczowym[2]. Odsetek powikłań był duży, a jakość życia wielu pacjentów z wyleczoną chirurgicznie wadą była obniżona przez nietrzymanie stolca. Ponadto szereg typów malformacji odbytu i odbytnicy nie nadawał się do leczenia z dojścia kroczowego. Jedni z najwybitniejszych chirurgów dziecięcych XX wieku, William Ladd i Robert Gross z Boston Children's Hospital badali anatomiczne relacje struktur miednicy u pacjentów z malformacjami odbytu i odbytnicy i opublikowali je w klasycznym podręczniku Grossa w 1953 roku[4]. Wiele jednak z ich obserwacji było jedynie spekulatywnych i zbyt uproszczonych[5]. W tym samym czasie istotnych obserwacji klinicznych dokonał Douglas Stephens z Melbourne, który przeprowadził autopsje noworodków z ALM. Jego prace były jednak wypaczone przez fakt, że dzieci z lżejszymi wadami przeżywały i nie były w ten sposób badane[5]. W 1953 roku Stephens zaproponował operację z dojścia strzałkowego w okolicy krzyżowej, uzupełnioną przez brzuszno-kroczową operację w razie konieczności[6]. Celem wykonania cięcia w okolicy krzyżowej było oszczędzenie domniemanego mięśnia łonowo-odbytniczego, co uznano za kluczowe dla zachowania czynności trzymania kału w przyszłości.
W 1969 roku dr Alberto Peña odbywający staż w Boston Children’s Hospital spotkał się ze współpracownikiem Stephensa, Justinem Kellym. Dr Kelly był autorem stosowanej w bostońskiej klinice techniki, polegającej na wykonaniu małego cięcia w okolicy krzyżowej i odpreparowaniem tkanek „na ślepo” w okolicy cewki moczowej by umożliwić przeciągnięcie odbytnicy przez pętlę mięśni łonowo-odbytniczych. W 1972 Peña wrócił do miasta Meksyk i jako ordynator tamtejszej kliniki chirurgicznej skupił swoje zainteresowanie na chirurgicznym leczeniu malformacji odbytu i odbytnicy. Zaczął operować pacjentów według sposobu zaproponowanego przez Stephensa, stopniowo zwiększając długość wykonanego cięcia, co pozwalało na lepsze uwidocznienie odbytnicy i przyległych tkanek. Gdy przedstawił swoje ośmioletnie doświadczenie kliniczne z 56 pacjentami na spotkaniu Pacific Association of Pediatric Surgeons w Colorado Springs, proponowane przez niego rozwiązania spotkały się ze sceptycznym przyjęciem. Peña zdecydował się po powrocie do miasta Meksyk na wykonanie operacji z dużego cięcia strzałkowego tylnego i przeprowadził pierwszy taki zabieg 12 sierpnia 1980[5]. Stwierdził wówczas, że postulowany mięsień łonowo-odbytniczy nie istnieje, a włókna mięśniowe tworzą raczej lejkowaty mechanizm zwieraczowy. W 1982 roku Peña opublikował wyniki swoich badań, z czasem metoda ta (określana niekiedy jako plastyka odbytu sposobem Peñy), jako lepsza i skuteczniejsza w leczeniu złożonych malformacji, stała się standardem leczenia tych wad. W przypadku wysokiego położenia odbytnicy i pochwy metodę łączy się z zabiegiem z dojścia przez jamę brzuszną (który może być wykonany metodą laparoskopową).
Epidemiologia
Częstość malformacji odbytu i odbytnicy szacuje się na 1:5000 żywych urodzeń, chociaż wyliczano również tak dużą częstość jak 1:1862[7] i tak niską jak 1:9630[8]. W Polsce, częstość urodzeń dzieci z takimi wadami w województwie łódzkim oszacowano na średnio 1:2295[9].
Etiologia


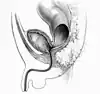

Malformacje okolicy odbytu i krocza powstają wskutek nieprawidłowego wytworzenia przegrody kloaki i kanału odbytu w embriogenezie. Przetrwała kloaka (cloaca persistens) powstaje wskutek całkowitego niezstąpienia przegrody moczowo-odbytniczej i otwarciu jelita pierwotnego i pochwy do wspólnej jamy. Zarośnięcie odbytowo-odbytnicze (atresia anorectalis) jest skutkiem nieprawidłowego wykształcenia dystalnego odcinka odbytnicy, a błoniaste zarośnięcie odbytu (atresia ani membranosa) – niewystąpienia perforacji błony odbytu około 7. tygodnia życia płodowego. Bardzo rzadka malformacja, jaką jest agenezja odbytnicy, jest postacią niedrożności jelita.
Klasyfikacja
Najprostszy i wciąż mający zastosowanie kliniczne podział z Melbourne (1979) to klasyfikacja malformacji odbytu i odbytnicy na dwa typy:
- typ niski (typ przezdźwigaczowy, translevator), w którym odbytnica przechodzi przez pętlę łonowo-odbytniczą,
- typ wysoki (typ naddźwigaczowy, supralevator), w którym odbytnica nie przechodzi przez pętlę łonowo-odbytniczą.
Wady pośrednie określa się w tej klasyfikacji jako te, w których odbytnica (także z przetoką) znajduje się w obrębie kompleksu mięśniowego dźwigaczy.
Bardziej złożone klasyfikacje zaproponowane znajdują się poniżej:
| Wady | Chłopcy | Dziewczynki |
| Wady wysokie | Atrezja odbytu z przetoką do części sterczowej cewki
Atrezja odbytu bez przetoki Atrezja odbytnicy |
Atrezja odbytu z przetoką odbytniczo-pochwową
Atrezja odbytu bez przetoki Atrezja odbytnicy |
| Wady pośrednie | Atrezja odbytu z przetoką odbytniczo-cewkowo-opuszkową Atrezja odbytu bez przetoki |
Atrezja odbytu z przetoką odbytniczo-przedsionkową Atrezja odbytu z przetoka odbytniczo-pochwową Atrezja odbytu bez przetoki |
| Wady niskie | Atrezja odbytu z przetoką odbytowo-skórną Zwężenie odbytu |
Atrezja odbytu z przetoką odbytowo-przedsionkową Atrezja odbytu z przetoką odbytowo-skórną Zwężenie odbytu |
| Wady stekowe | wysokie pośrednie niskie |
wysokie pośrednie niskie |
| Płeć męska | Przetoka odbytniczo-kroczowa
Przetoka odbytniczo-cewkowo-opuszkowa |
| Płeć żeńska | Przetoka odbytniczo-kroczowa
Przetoka odbytniczo-przedsionkowa |
| Złożone i rzadkie wady | Wynicowanie kloaki
Tylna kloaka |
Malformacje odbytu i odbytnicy mogą być izolowanymi wadami albo wchodzą w skład zespołów i asocjacji wad wrodzonych; niektóre z nich to[2]:
- asocjacja VACTERL
- asocjacja MURCS
- asocjacja OEIS
- osiowa dysplazja mezodermalna
- zespół Klippla-Feila
- zespół regresji kaudalnej i syrenomelia
- zespół Downa
- zespół Pataua
- zespół Edwardsa
- trisomia 16
- delecja długich ramion chromosomu 13
- zespół Pallistera-Killiana
- zespół kociego oka
- rodzicielska unidisomia 16
- zespół delecji 22q11
- zespół Currarino
- zespół Pallistera i Hall
- zespół Townesa i Brocksa
- zespół łokciowo-sutkowy
- zespół Okihiro (OMIM#607323)
- zespół Riegera (OMIM#180500, OMIM%601499)
- dysplazja śmiertelna
- choroba Hirschsprunga
- zespół Feingolda
- zespół Kabuki
- zespół Opitza-G
- zespół Johnsona-Blizzarda (OMIM#243800)
- dyzostoza kręgowo-żebrowa (OMIM#277300, OMIM#608681, OMIM#609813)
- zespół krótkie żebro-polidaktylia
- zespół Ballera-Gerolda (OMIM#218600)
- ciliopatie
- zespół Frasera (OMIM#219000)
- zespół Lowe’a
- heterotaksja (OMIM#306955)
- zespół FG
- opóźnienie umysłowe związane z chromosomem X
- zespół MIDAS
- zespół Christiana.
Typy wad u chłopców
- Przetoka odbytniczo-kroczowa
W wadzie tej najniższa część odbytnicy otwiera się przetoką na kroczu proksymalnie od środka mięśnia zwieracza zewnętrznego odbytu; zwieracz otacza odbytnicę. Jeśli przetoka nie ma ujścia, pod skórą krocza w pobliżu szwu moszny może przeświecać smółka. Nierzadko stwierdza się przetokę pokrytą fałdem skórnym („rączka koszyka”, ang. bucket handle).
- Przetoka odbytniczo-cewkowa
- Przetoka odbytniczo-pęcherzowa
- Atrezja odbytu bez przetoki
- Atrezja lub zwężenie odbytnicy
Typy wad u dziewczynek
- Przetoka odbytniczo-kroczowa
- Przetoka odbytniczo-przedsionkowa
- Atrezja odbytu bez przetoki
- Atrezja lub zwężenie odbytnicy
- Przetrwały stek
Towarzyszące wady
- Wady kręgosłupa i rdzenia kręgowego
U pacjentów z malformacjami odbytu i odbytnicy najczęściej spotyka się nieprawidłowości budowy kości krzyżowej. W celu określenia stopnia hipoplazji kości krzyżowej najczęściej liczone są trzony kręgów krzyżowych; bardziej obiektywną metodą oceny jest obliczenie indeksu krzyżowego (sacral ratio) przez zmierzenie długości kości krzyżowej i porównanie jej z wymiarami miednicy. Zdjęcie boczne jest zalecane, ponieważ na zdjęciu przednio-tylnym pomiar jest zniekształcony przez nachylenie miednicy[2]. Wadom typu połowiczej kości krzyżowej (hemisacrum) zawsze towarzyszy masa w okolicy przedkrzyżowej, najczęściej rozpoznawana jako torbiel skórzasta, potworniak, tłuszczakooponiak albo przednia przepuklina oponowa. Półkręgi mogą występować też w odcinku lędźwiowym i piersiowym kręgosłupa, prowadząc do skoliozy. Inne wady układu szkieletowego to bloki kostne, brak kości ogonowej.
Mogą współistnieć wady rdzenia kręgowego, w tym zespół zakotwiczenia rdzenia kręgowego[10][11]. Dochodzi do niego wskutek uwięźnięcia nici końcowej rdzenia między kręgami; częstość występowania szacuje się na około 25% pacjentów z ARM, a ryzyko jest tym większe im malformacja jest „wyższa” i bardziej złożona. Większe ryzyko zespołu dotyczy też pacjentów z niedorozwojem kości krzyżowej i towarzyszącymi wadami układu moczowego[2]. Objawy mogą polegać na zaburzeniach czuciowych i ruchowych kończyn dolnych; czynnościowe rokowanie w tej grupie pacjentów jest gorsze, pacjenci z ANM i zespołem zakotwiczenia rdzenia kręgowego mają też wyższe wady, gorzej rozwinięte kości krzyżowe, towarzyszące wady rdzenia i gorzej rozwinięte mięśnie krocza; rzeczywisty wpływ samego zespołu zakotwiczenia na rokowanie jest więc niejasny. W literaturze neurochirurgicznej zaleca się uwolnienie rdzenia, ale nie stwierdzono wpływu zabiegu na rokowanie u pacjentów z ANM[2]. USG rdzenia w pierwszych trzech miesiącach życia i późniejsze badanie MRI pozwalają ustalić rozpoznanie. Inne wady rdzenia stwierdzane u pacjentów z ANM to jamistość rdzenia i przepuklina oponowo-rdzeniowa.
- Wady układu moczowo-płciowego
Wady układu moczowo-płciowego występują u do 50% wszystkich pacjentów z malformacjami odbytu i odbytnicy[2]. Niektóre z nich to agenezja nerki, dysplazja nerki, nerka podkowiasta, odpływ pęcherzowo-moczowodowy, wodonercze, spodziectwo, moszna dwudzielna. Wszystkie dzieci z tymi wadami muszą być przebadane po urodzeniu w celu wykluczenia wad z tej grupy, a najcenniejszym badaniem jest USG jamy brzusznej i miednicy. Badanie układu moczowego przed wykonaniem zabiegu kolostomii dostarcza chirurgowi informacji niezbędnych do rozwiązania podczas zabiegu ewentualnych problemów urologicznych. Częstym błędem jest rozpoznawanie przy badaniu krocza dzieci płci żeńskiej atrezji odbytu z przetoką odbytniczo-pochwową, podczas gdy drogi moczowe, pochwa i odbytnica dziecka tworzą wspólny kanał określany jako kloaka[10][12][13]. Obecność pojedynczego otworu w kroczu jest objawem przetrwałej kloaki. Pacjenci z tą wadą mają też małe narządy płciowe zewnętrzne. U pacjentek z kloaką, badaniem palpacyjnym jamy brzusznej można wyczuć guzowatą zmianę, będącą w rzeczywistości poszerzoną pochwą (hydrocolpos); objaw ten stwierdzany jest u 50% pacjentek z kloaką. USG jamy brzusznej pozwala stwierdzić obecność uropatii zaporowej i poszerzonej pochwy. Nieprawidłowe rozpoznanie ma istotne implikacje terapeutyczne. Zasadniczą kwestią jest prawidłowe rozpoznanie kloaki, jako że u 90% dzieci występuje towarzysząca wada układu moczowego, a u 50% stwierdza się hydrocolpos. Obydwa stany powinny być właściwie rozpoznane w okresie noworodkowym, co pozwala uniknąć poważnych powikłań. Nierozpoznanie kloaki zwykle skutkuje przeoczeniem uropatii zaporowej, a pacjent podczas zabiegu operacyjnego może mieć przeprowadzoną jedynie kolostomię; następnie mogą wystąpić takie powikłania jak sepsa, kwasica i niekiedy śmierć. Ponadto zwykle udaje się wówczas skorygować chirurgicznie jedynie odbytniczą komponentę złożonej wady, z pozostawieniem nieskorygowanej zatoki moczowo-płciowej[2].
Rozpoznanie


Rozpoznanie atrezji odbytu stawiane jest zazwyczaj natychmiast po urodzeniu wobec braku odbytu skórnego. Radiologicznie określa się długość zarośniętego odcinka odbytnicy. Najczęściej wykonuje się w tym celu zdjęcie rentgenowskie uwidaczniające, do jakiej wysokości dochodzi powietrze w obrębie odbytnicy. Robi się wówczas zdjęcie boczne, a noworodek znajduje się w pozycji głową w dół. Metodą alternatywną jest ultrasonografia przezkroczowa. U dzieci starszych metodą z wyboru jest tomografia rezonansu magnetycznego. Stwierdzenie atrezji odbytu jest wskazaniem do USG jamy brzusznej i cystoureterografii mikcyjnej (poszukiwanie wad układu moczowego).
- Badanie kliniczne
Malformacje odbytu i odbytnicy powinny być rozpoznane we wstępnym badaniu klinicznym noworodka po urodzeniu. Uważne oglądanie krocza pozwala na stwierdzenie braku odbytu skórnego, zagłębienia odbytu z charakterystycznym fałdem skórnym albo przetoki odbytniczo-skórnej. Stwierdzenie płaskiego krocza bez zaznaczonego zagłębienia międzypośladkowego sugeruje wadę wysoką. U dziewczynek dokładne obejrzenie krocza i przedsionka pochwy pozwala nierzadko określić rodzaj wady; badanie przedsionka pochwy może wymagać użycia cienkiego cewnika, którym można stwierdzić obecność ukrytej pod fałdem błony śluzowej przetoki odbytniczo-przedsionkowej. Brak tylnego brzegu błony dziewiczej występuje w rzadkiej postaci wady jaką jest przetoka odbytniczo-pochwowa. Pojedynczy otwór w hipoplastycznym sromie z którego wydostają się mocz i smółka to obraz wspólnego kanału przetrwałego steku; współwystępowanie wyczuwalnego przez powłoki skórne brzucha guza sugeruje rozpoznanie hydrocolopos lub megavesica. Wady niskie u dziewczynek mają postać otworu przetoki w linii środkowej krocza lub w obrębie skóry spoidła tylnego warg sromowych. Dodatkowymi informacjami pozwalającymi na postawienie rozpoznania są obecność smółki w moczu lub zabrudzenie smółką sromu, wskazujące jednoznacznie na istnienie przetoki.
- Radiogram przeglądowy
Na przeglądowym rtg jamy brzusznej dziecka można stwierdzić cień gazu w pęcherzu w przypadku obecności przetoki do dróg moczowych. Dokładne określenie relacji anatomicznych narządów miednicy mniejszej nie jest możliwe.
- Zdjęcie rtg w pozycji bocznej odwróconej (inwertogram)
Badanie to, podobnie jak zdjęcie w pozycji leżącej na brzuchu z uniesioną miednicą promieniem bocznym pozwala na zlokalizowanie ślepego końca odbytnicy względem zaznaczonego na skórze krocza miejsca spodziewanego odbytu. Gaz w ślepym końcu odbytnicy pojawia się najwcześniej 18 godzin od narodzin dziecka, i dopiero po upływie tego czasu wykonanie radiogramu jest celowe.
- USG jamy brzusznej
USG jamy brzusznej jest wskazane po rozpoznaniu malformacji odbytu i odbytnicy; ma na celu wykluczenie ewentualnych wad nerek lub wodonercza.
- USG przezkroczowe

Wykonywane po rozpoznaniu wady, ma na celu wykluczenie współistniejących wad serca i wielkich naczyń.
- Kolostogram
Badanie kontrastowe obwodowego, wyłączonego przez kolostomię odcinka jelita, poprzedzające zabieg operacyjny. Pozwala na precyzyjne określenie rodzaju wady, obecności i lokalizacji przetoki oraz poziomu niedrożności (Niedzielski, 1998, Shaul, 1997). Badanie polega na podaniu wodnego roztworu środka kontrastowego do przetoki jelita za pomocą cewnika Foleya z balonem wypełnionym poniżej stomii i wykonaniu zdjęcia rtg w pozycji bocznej. Środek cieniujący podawany jest automatyczną strzykawką pod odpowiednim ciśnieniem pod kontrolą fluoroskopii. W przypadku złożonych wad (przetrwały stek) konieczne jest podanie środka kontrastowego dodatkowo przez pojedynczy otwór w sromie.
Leczenie
Postępowanie pooperacyjne
Tuż przed operacją wdraża się antybiotykoterapię, do trzech dni po zabiegu. Cewnik Foleya utrzymywany jest w pęcherzu chłopców leczonych z powodu przetoki odbytniczo-cewkowej lub odbytniczo-pęcherzowej przez 5-7 dni. Operacja przetrwałej kloaki wymaga dłuższego utrzymywania cewnika w miejscu, do 14 dni. Po 14-20 dniach od operacji rozpoczyna się rozszerzanie odbytu; pierwszy zabieg wykonuje chirurg, następne mogą wykonywać opiekunowie dziecka. Odbyt poszerzany jest dwa razy dziennie za pomocą mechanicznego rozszerzadła Hegara, którego średnicę zwiększa się o 1 mm w cotygodniowych odstępach. Po uzyskaniu oczekiwanej średnicy odbytu zamyka się operacyjnie kolostomię, poszerzanie kontynuując przez następne trzy miesiące.
W okresie noworodkowym chirurg musi zadecydować, czy dziecko wymaga przeprowadzenia kolostomii czy też można przeprowadzić operację korekcji chirurgicznej wady.
Kolostomia
Celem kolostomii jest ograniczenie zakażeń dróg moczowych za pośrednictwem przetoki i zmniejszenie rozdęcia obwodowego odcinka jelita grubego, a także uniknięcie powikłań późniejszej zasadniczej operacji wytwórczej odbytu. Kolostomia daje też możliwość wykonania badań obrazowych dających wgląd w typ wady i pozwala na dokładne zaplanowanie zabiegu operacyjnego. Preferowana jest kolostomia dwulufowa w obrębie zstępnicy i esicy, wykonywana ze skośnego nacięcia w dolnym lewym kwadrancie brzucha.
Operacja z tylnego dojścia strzałkowego
Operacja z tylnego dojścia strzałkowego sposobem Peñy (posterior sagittal anorectoplasty, PSARP) jest obecnie złotym standardem leczenia operacyjnego malformacji odbytu i odbytnicy. Technika ta pozwala na racjonalne wyleczenie dzięki lepszemu wglądowi w anatomię danej wady, i uniknięcie uszkodzenia ważnych struktur anatomicznych: nasieniowodu, pęcherzyków nasiennych, gruczołu krokowego, ektopowo przebiegającego moczowodu. Podczas operacji stosuje się stymulator elektryczny, co pozwala na rozpoznanie i precyzyjne rozdzielenie włókien mięśniowych zwieracza zewnętrznego i dźwigaczy.
- Przetoka odbytniczo-pęcherzowa
W rzadkich przypadkach tej naddźwigaczowej malformacji zalecaną metodą leczenia chirurgicznego wady jest połączenie operacji z tylego dojścia strzałkowego i operacji brzusznej, metodą laparoskopową lub na drodze laparotomii.
- Atrezja odbytu bez przetoki
U pacjentów z atrezją odbytu bez przetoki wymagane jest cięcie wykonane w taki sam sposób, pozwalające na precyzyjne odpreparowanie dystalnego odcinka odbytnicy od dróg moczowych.
- Przetoka odbytniczo-przedsionkowa
W przypadkach z przetoką między odbytnicą a przedsionkiem pochwy tylne cięcie strzałkowe krocza może być krótsze niż u chłopców z przetoką odbytniczo-cewkową. Często cały kompleks dźwigacza odbytu nie musi być rozdzielany i jedynie zwieracz zewnętrzny odbytu i dolna część dźwigacza muszą być oddzielone. W tym typie malformacji wspólne są ściana przednia odbytnicy i tylna ściana pochwy, a ich rozdzielenie jest najtrudniejszą częścią operacji. Kiedy odbytnica zostanie całkowicie uwolniona, wykonuje się rekonstrukcję krocza i odbytnicę umieszcza się w ograniczeniu kompleksu zwieraczy.
- Atrezja odbytnicy
dopracować
- Przetrwała kloaka
Operacja wspierana laparoskopowo
Operacja z dojścia strzałkowego przedniego wspierana laparoskopowo. Odbytnica jest oswobadzana, a następnie przeprowadzana przez dno miednicy mniejszej i zwieracze drogą minimalnego cięcia przezkroczowego. Cięcie przezkroczowe dokonane pod źródłem światła laparoskopu umożliwia precyzyjniejsze umieszczenie trokaru i przeprowadzenie odbytnicy przez kompleks mięśnia zwieracza zewnętrznego odbytu. Operacja ta może być wykonana zarówno w okresie noworodkowym bez wcześniejszej kolostomii lub później, po wcześniejszej kolostomii, jako kolejny etap naprawy wady. Ta nowa technika wprowadzona przez Keitha E. Georgesona i wsp. wymaga długoterminowych badań klinicznych w celu określenia odległych skutków operacji[15][16].
Operacja z przedniego dojścia strzałkowego
Operacja z przedniego dojścia strzałkowego wykonywana jest z przedniego strzałkowego cięcia krocza (od podstawy moszny do tylnej części środka ścięgnistego krocza), co pozwala zachować nienaruszoną mięśniówkę wewnętrznego zwieracza odbytu. Istnieje ryzyko uszkodzenia splotu żylnego pęcherzykowego podczas resekowania połączenia przetoki z cewką moczową.
Powikłania

Powikłania PSARP można podzielić na wczesne i późne. Powikłaniem nie jest upośledzenie czynności wytworzonego odbytu. Wczesnymi komplikacjami są: zakażenie i rozejście rany, zakażenie i rozejście zespolenia, niedokrwienie jelita, neurogenny pęcherz moczowy, śródoperacyjne uszkodzenie cewki moczowej, nasieniowodu lub pochwy. Późnymi powikłaniami są zwężenie wytworzonego odbytu, zwężenie pochwy, wytworzenie przetoki odbytniczo-cewkowej, wypadanie błony śluzowej odbytnicy.
Klasyfikacja ICD10
| kod ICD10 | nazwa choroby |
|---|---|
| ICD-10: Q42 | Wrodzony brak, zarośnięcie lub zwężenie jelita grubego[17] |
| ICD-10: Q42.0 | Wrodzony brak, zarośnięcie lub zwężenie odbytnicy z przetoką |
| ICD-10: Q42.1 | Wrodzony brak, zarośnięcie lub zwężenie odbytnicy bez przetoki |
| ICD-10: Q42.2 | Wrodzony brak, zarośnięcie lub zwężenie odbytu z przetoką |
| ICD-10: Q42.3 | Wrodzony brak, zarośnięcie lub zwężenie odbytu bez przetoki |
| ICD-10: Q42.8 | Wrodzony brak, zarośnięcie lub zwężenie innych części jelita grubego |
| ICD-10: Q42.9 | Wrodzony brak, zarośnięcie lub zwężenie jelita grubego, w części nie określonej |
Przypisy
- ↑ Obejmuje: wrodzoną niedrożność, zamknięcie lub zwężenie jelita grubego
- 1 2 3 4 5 6 7 8 9 10 Levitt, MA, Peña, A. Anorectal malformations. „Orphanet Journal of Rare Diseases”. 2. 33, 2007. 17651510.
- ↑ Amusset, JZ. Observation sur une opération d’anus artificiel par un nouveau procédé à la region anale d’un enfant nouveau-né, dans un cas d’absence congéntitale du rectum, suivie de quelques réflexions sur les obturations du gros intestin. „Annales d'hygiène publique et de médecine légale”, 1835.
- ↑ Gross RE: The surgery of infancy and chilhood. Philadelphia: WB Saunders, 1953.
- 1 2 3 Colorectal Center for Children: History of the Treatment of Anorectal Malformations. Imperforate Anus and Cloaca: Anorectal Malformations, 2003. [dostęp 2007-09-21]. [zarchiwizowane z tego adresu (2004-12-23)]. (ang.).
- ↑ Stephens FD. Malformations of the anus. „Aust N Z J Surg”. 23. 1, s. 9-24, 1953. PMID: 13081533.
- ↑ A surgeon's experience. W: Chatterjee SK: Anorectal malformations. New York: Oxford University Press, 1993.
- ↑ Ivy RH. Congenital abnormalities. „Plast Reconstr Surg”. 20, s. 400, 1957.
- ↑ Niedzielski J. Incidence of anorectal malformations in Łódź province. „Med Sci Monit”. 6. 1, s. 133-136, 2000. PMID: 11208300.
- 1 2 Levitt MA, Patel M, Rodriguez G, Gaylin DS, Pena A. The tethered spinal cord in patients with anorectal malformations. „J Pediatr Surg”. 32. 3, s. 462-468, 1997. PMID: 9094019.
- ↑ Tuuha SE, Aziz D, Drake J, Wales P, Kim PC. Is surgery necessary for asymptomatic tethered cord in anorectal malformation patients?. „J Pediatr Surg”. 39. 5, s. 773-777, 2004. PMID: 15137017.
- ↑ Levitt MA, Pena A. Pitfalls in the management of newborn cloacas. „Pediatr Surg Int”. 21. 4, s. 264-269, 2005. PMID: 15726389.
- ↑ Rosen NG, Hong AR, Soffer SZ, Rodriguez G, Pena A. Rectovaginal fistula: a common diagnostic error with significant consequences in girls with anorectal malformations. „J Pediatr Surg”. 37. 7, s. 961-965, 2002. PMID: 12077749.
- 1 2 Pratap A, Tiwari A, Kumar A, Adhikary S, Singh SN, Paudel BH, Bartaula R, Mishra B. Sphincter saving anorectoplasty (SSARP) for the reconstruction of Anorectal malformations. „BMC Surg”. 7. 20, 2007. DOI: 10.1186/1471-2482-7-20. PMID: 17892560.
- ↑ Sydorak RM, Albanese CT. Laparoscopic repair of high imperforate anus. „Semin Pediatr Surg”. 11. 4, s. 217-225, 2002. PMID: 12407503.
- ↑ Georgeson KE, Inge TH, Albanese CT. Laparoscopically assisted anorectal pull-through for high imperforate anus – a new technique. „J Pediatr Surg”. 35. 6, s. 927-930, 2000. PMID: 10873037.
- ↑ Obejmuje: wrodzoną niedrożność, zamknięcie lub zwężenie jelita grubego
Bibliografia
- Wady odbytu i odbytnicy. W: Elżbieta Grochowska: Chirurgia dziecięca. Jerzy Czernik (red.). Wyd. Wydanie I. Warszawa: Wydawnictwo Lekarskie PZWL, 2005, s. 181-200. ISBN 83-200-3066-8.
- Marc A Levitt, Alberto Peña. Anorectal malformations. „Orphanet Journal of Rare Diseases”. 2. 33, 2007. PMID: 17651510.
- Lech Korniszewski: Dziecko z zespołem wad wrodzonych. Diagnostyka dysmorfologiczna. Wyd. Wydanie II. Wydawnictwo Lekarskie PZWL, 2005, s. 119-121. ISBN 83-200-3042-0.
- Chapter 27. Anorectal Anomalies. W: Alberto Peña, Marc A. Levitt: Pediatric Surgery. P. Puri, M. E. Höllwarth (Eds.). Berlin-Heidelberg-New York: Springer, s. 289-312. ISBN 3-540-40738-3.
- Pratap A, Tiwari A, Kumar A, Adhikary S, Singh SN, Paudel BH, Bartaula R, Mishra B. Sphincter saving anorectoplasty (SSARP) for the reconstruction of Anorectal malformations. „BMC Surg”. 7. 20, 2007. DOI: 10.1186/1471-2482-7-20. PMID: 17892560.
Linki zewnętrzne
- Nelson G Rosen, Daniel A Beals, David Rothstein: Imperforate Anus. eMedicine Pediatrics, 2007. [dostęp 2007-09-15]. (ang.).
- Marc A Levitt, Alberto Pena: Imperforate Anus: Surgical Perspective. eMedicine Pediatrics, 2007. [dostęp 2007-09-21]. (ang.).
- Colorectal Center for Children: History of the Treatment of Anorectal Malformations. Imperforate Anus and Cloaca: Anorectal Malformations, 2003. [dostęp 2007-09-21]. [zarchiwizowane z tego adresu (2004-12-23)]. (ang.).
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.