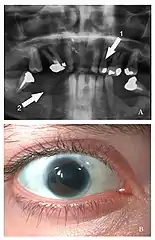
 Cechy fenotypowe zespołu Axenfelda i Riegera u 21-letniej pacjentki. Na pantomogramie zaznaczono hipodoncję (strzałka 1) i obszar hipoplazji szczęki (strzałka 2); na zdjęciu niżej widać dodatkowy otwór źreniczny (polycoria) | |
| Klasyfikacje | |
| ICD-10 |
Q13.8 |
|---|---|
| DiseasesDB | |
Zespół Axenfelda-Riegera (ang. Axenfeld-Rieger syndrome, ARS) – zespół wad wrodzonych, charakteryzujący się występowaniem obustronnych wad przedniego odcinka oka skojarzonych z wadami rozwojowymi zębów, środkowej części twarzy i jamy brzusznej. Większość przypadków zespołu wiąże się z mutacjami genów PITX2 lub FOXC1. Zespół opisali niezależnie od siebie Theodor Axenfeld i Herwigh Rieger.
Obraz kliniczny
W spektrum klinicznym zespołu Axenfelda-Riegera zawierają się, początkowo opisywane osobno, anomalia Axenfelda i anomalia Riegera. Ponadto osobno traktowano tzw. zespół Riegera:
- anomalia Axenfelda jest zaburzeniem budowy kąta przesączania, w którym powstają zrosty między przesuniętą do przodu, nierówną i wystającą linią Schwalbego a pasmami odchodzącymi od obwodowej części tęczówki. Jaskra wrodzona wtórna w anomalii Axenfelda występuje dosyć rzadko;
- anomalia Riegera polega na dysgenezji tęczówkowo-rogówkowej z wyraźną hipoplazją zrębu tęczówki, przemieszczeniem źrenicy (corectopia) i pełnościennymi otworami tęczówki (pseudopolycoria). Linia Schwalbego w anomalii Axenfelda jest niekiedy przemieszczona do komory przedniej (ang. posterior embryotoxon). W 50-60% przypadków w wyniku dysgenezji rozwija się jaskra wtórna wrodzona;
- w zespole Riegera obok obustronnych wad przedniego odcinka oka stwierdza się dodatkowo wady zębów, takie jak mikrodoncja, hipodoncja, anodoncja albo hipoplazja szczęki, cechy dysmorficzne twarzy: szeroki grzbiet nosa, telekantus, hiperteloryzm oczny.
Etiologia
ARS powiązano z pięcioma loci genowymi (4q25, 6p25, 11p13, 13q14, 16q24). Stwierdzono mutacje w trzech genach kodujących czynniki transkrypcyjne; dwie z nich - dotyczące genów PITX2 i FOXC1 w loci, odpowiednio, 4q25 i 6p25, są najczęstsze. W jednym przypadku stwierdzono mutację w genie PAX6 w locus 11p13.
Bibliografia
- de la Houssaye G, Bieche I, Roche O, Vieira V, Laurendeau I, Arbogast L, Zeghidi H, Rapp P, Halimi P, Vidaud M, Dufier JL, Menasche M, Abitbol M. Identification of the first intragenic deletion of the PITX2 gene causing an Axenfeld-Rieger Syndrome: case report. „BMC Med Genet”. 7. 82, 2006. DOI: 10.1186/1471-2350-7-82. PMID: 17134502.
- Lines MA, Kozlowski K, Walter MA. Molecular genetics of Axenfeld-Rieger malformations. „Hum Mol Genet”. 11. 10, s. 1177-84, 2002. PMID: 12015277.
- Okulistyka. Podstawy kliniczne. Maria Hanna Niżankowska (red.). Warszawa: Wydawnictwo Lekarskie PZWL, 2007, s. 366. ISBN 978-83-200-3224-6.
Linki zewnętrzne
- RIEGER SYNDROME, TYPE 1; RIEG1 w bazie Online Mendelian Inheritance in Man (ang.)
- Axenfeld's syndrome w bazie Who Named It (ang.)
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.