
Metabolizm (z gr. μεταβολή 'zmiana' od μετά 'ponad, poza' i βάλλειν 'rzut') – całokształt reakcji chemicznych i związanych z nimi przemian energii zachodzących w żywych komórkach, stanowiący podstawę wszelkich zjawisk biologicznych. Procesy te pozwalają komórce na wzrost i rozmnażanie, zarządzanie swoją strukturą wewnętrzną oraz odpowiadanie na bodźce zewnętrzne.
Reakcje chemiczne składające się na metabolizm są zorganizowane w szlaki metaboliczne. Są to szeregi reakcji, w których produkty jednej reakcji (nazywane tu metabolitami) są używane jako substraty kolejnej reakcji, a w przekształceniach tych zwykle udział biorą enzymy. Enzymy pozwalają na przeprowadzenie reakcji, które w praktyce nie zaszłyby bez ich udziału, ponieważ byłyby termodynamicznie niekorzystne. Ich działanie polega na obniżaniu energii aktywacji i zwiększaniu szybkości reakcji, sprzęganiu ich z reakcjami spontanicznymi wyzwalającymi energię (korzystnymi termodynamicznie). Enzymy pozwalają na regulację szlaków metabolicznych w odpowiedzi na zmiany warunków wewnątrz komórki lub sygnały pochodzące spoza komórki.
Szlaki metaboliczne można podzielić na dwie duże klasy: przekształcające związki chemiczne z wytworzeniem energii w postaci użytecznej biologicznie oraz wymagające dostarczenia energii, aby mogły zachodzić[1]. Pierwsze z nich, będące reakcjami egzoenergetycznymi, w czasie których następuje przekształcanie związków organicznych w energię, nazywa się reakcjami katabolicznymi lub bardziej ogólnie katabolizmem. Drugie natomiast, będące reakcjami endoenergetycznymi, czyli wymagającymi dostarczenia energii, jak tworzenie glukozy, lipidów lub białek, nazywa się reakcjami anabolicznymi lub anabolizmem[1][2].
Genetycznie uwarunkowane możliwości metaboliczne danego organizmu decydują o zakwalifikowaniu danej substancji jako „przydatnej” lub „nieprzydatnej” (lub nawet „trującej”), jej użyciu i przetworzeniu. Dla przykładu, niektóre organizmy prokariotyczne (np. bakterie z rodzaju Beggiatoa) używają siarkowodoru jako źródła energii, włączając go w swoje szlaki metaboliczne, podczas gdy m.in. dla zwierząt gaz ten jest trujący[3] (H
2S blokuje oksydazę cytochromową[4]). Tempo metabolizmu wpływa natomiast na ilość pożywienia, jaka będzie niezbędna do prawidłowego funkcjonowania danego organizmu.
Szlaki metaboliczne wykazują duże podobieństwo nawet u gatunków o niezwykle dalekim pokrewieństwie. Przykładowo zestaw enzymów, tożsamych w funkcji i niezwykle podobnych w strukturze, biorących udział w cyklu kwasu cytrynowego można znaleźć zarówno u bakterii Escherichia coli, jak i u organizmów wielokomórkowych[5]. Ta uniwersalność szlaków metabolicznych jest prawdopodobnie efektem ich dużej wydajności, a więc istniejącej, dodatniej presji ewolucyjnej do ich podtrzymania, a także wczesnego pojawienia się w ewolucyjnej historii życia[6][7].
Podstawowe substancje

Większość struktur tworzących ciała zwierząt, roślin i innych żywych organizmów zbudowana jest z trzech podstawowych typów związków: aminokwasów, węglowodanów oraz lipidów. Podstawowe związki (np. aminokwasy) mogą łączyć się w polimery kondensacyjne, tworząc wyżej zorganizowane cząsteczki (np. białka). Polimerami kondensacyjnymi są także kwasy nukleinowe, jednak cząsteczki budujące je – nukleotydy składają się z kilku prostszych związków chemicznych. Jako że wymienione podstawowe typy związków są niezbędne dla życia, w procesach anabolicznych organizm zajmuje się ich syntezą podczas budowy swoich komórek oraz – w przypadku pożywienia – katabolicznym rozkładem i wykorzystaniem uwolnionej energii lub ewentualnie pozyskiwaniem prostszych związków na drodze rozkładu bardziej złożonych. Makrocząsteczki te stanowią składnik każdego żywego organizmu. Niektóre z nich przedstawione są w poniższej tabeli.
| Typ cząsteczki | Nazwa formy monomerycznej | Nazwa formy polimerycznej | Przykłady form polimerycznych |
|---|---|---|---|
| Aminokwasy | Aminokwasy | Białka (lub polipeptydy) | Białka fibrylarne i globuliny |
| Węglowodany | Monosacharydy | Polisacharydy | Skrobia, glikogen i celuloza |
| Kwasy nukleinowe | Nukleotydy | Polinukleotydy | DNA i RNA |
Aminokwasy i białka
Białka zbudowane są z aminokwasów, połączonych liniowo wiązaniami peptydowymi. Wiele białek to enzymy katalizujące reakcje chemiczne metabolizmu. Inne pełnią funkcje strukturalne i mechaniczne, na przykład budują cytoszkielet warunkujący kształt komórki[8]. Są również ważnym elementem procesów takich, jak sygnalizacja komórkowa, odpowiedź immunologiczna, adhezja komórkowa, transport aktywny przez błony, cykl komórkowy i wiele innych[9]. Przebieg większości procesów komórkowych regulowany jest przez białka.
Lipidy
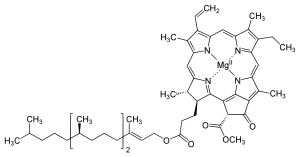
Lipidy to bardzo zróżnicowana grupa substancji biochemicznych. Definiowane są jako hydrofobowe lub amfifilowe cząsteczki o znaczeniu biologicznym, rozpuszczalne w rozpuszczalnikach organicznych (na przykład benzenie czy chloroformie)[10]. Lipidy (u eukariota głównie fosfolipidy) budują błony biologiczne oraz są jednym ze związków magazynujących energię[9]. Grupę lipidów stanowiąca związki zapasowe zwyczajowo określa się nazwą tłuszcze, są one zbudowanych z kwasów tłuszczowych oraz glicerolu. Cząsteczka glicerolu może być połączona z trzema cząsteczkami kwasów tłuszczowych[11]. W praktyce istnieje kilka wersji tej podstawowej struktury, zawierających na przykład dodatkowe grupy funkcyjne, takie jak fosforany w fosfolipidach. Inna klasą lipidów, do których należy m.in. cholesterol czy estrogen są steroidy, stanowiące kolejną dużą grupę lipidów produkowanych przez komórkę[12].
Węglowodany

Węglowodany to nierozgałęzione ketony lub aldehydy podstawione wieloma grupami hydroksylowymi i występujące w postaci liniowej lub pierścieniowej. Należą do najbardziej rozpowszechnionych substancji organicznych i spełniają w organizmach wiele funkcji, m.in. przechowywania i transportu energii (skrobia i glikogen), budowy struktur komórkowych (celuloza u roślin, chityna u zwierząt)[9]. Podstawowe monomery węglowodanowe, takie jak galaktoza, fruktoza i – najpopularniejsza – glukoza nazywane są monosacharydami. Mogą one łączyć się ze sobą na wyjątkowo wiele sposobów, tworząc polisacharydy[13].
Nukleotydy
Polimery kondensacyjne zwane DNA i RNA to długie łańcuchy zbudowane z nukleotydów. Cząsteczki te są niezbędne dla przechowywania i wykorzystywania informacji genetycznych, dzięki procesom transkrypcji i biosyntezy białek[9]. Informacje te są chronione przez mechanizmy naprawy DNA i powielane w procesie replikacji. Genom większości organizmów zapisany jest w postaci cząsteczek DNA, jednak niektóre wirusy zwane retrowirusami przechowują informację genetyczną w nici RNA. Przykładem retrowirusa jest wirus HIV, który używa enzymu odwrotnej transkryptazy, aby utworzyć kopię DNA ze swojego genomu RNA[14]. RNA, podobnie jak enzymy, może posiadać właściwości katalityczne i wtedy określa się je jako rybozymy, które wchodzą w skład spliceosomów czy rybosomów. Poszczególne nukleozydy powstają podczas dołączania cukru rybozy do właściwej zasady heterocyklicznej. Zasady te to związki zwane purynami i pirymidynami. W skład nukleotydów budujących RNA wchodzą adenina (A), uracyl (U), cytozyna (C) i guanina (G). W cząsteczkach DNA zamiast uracylu występuje tymina – T. Nukleotydy często pełnią też rolę koenzymów w reakcjach przenoszenia grup funkcyjnych[15].
Koenzymy

Metabolizm składa się z reakcji różnego typu, większość z nich jednak można zakwalifikować do kilku podstawowych grup ze względu na rodzaj przenoszonej grupy funkcyjnej[16]. Pozwoliło to komórkom na wykształcenie odpowiednich elementów metabolizmu odpowiedzialnych właśnie za przenoszenie tych grup pomiędzy różnymi związkami[15]. Są one zwane koenzymami. Każdemu rodzajowi reakcji enzymatycznej przyporządkowany jest określony koenzym; w komórce trwa nieprzerwanie proces tworzenia ich, więc po pewnym czasie są one rozkładane i wykorzystywane ponownie przez odpowiednie enzymy[17].
Przykładowym koenzymem jest adenozyno-5′-trifosforan (ATP), główny nośnik energii chemicznej w komórkach (oprócz niego w pewnych reakcjach zadanie to wypełniają analogiczne nukleotydy: GTP, UTP, CTP). Nukleotyd ten używany jest do przenoszenia energii chemicznej pomiędzy poszczególnymi reakcjami. Komórki zawierają stosunkowo niewielką ilość ATP, ale zapasy tego związku są nieprzerwanie odnawiane, toteż organizm ludzki zużywa w ciągu doby ilość ATP odpowiadającą masie jego ciała[17]. ATP stanowi łącznik pomiędzy katabolizmem i anabolizmem, jako że reakcje kataboliczne generują jego cząsteczki, zaś reakcje anaboliczne rozkładają je do adenozynodifosforanu (ADP). Związek ten pełni także funkcję nośnika reszt fosforanowych w reakcjach fosforylacji.
Witaminy to związki organiczne potrzebne organizmowi do prawidłowego funkcjonowania, jednak niemożliwe do wytworzenia w komórkach. U człowieka wiele witamin rozpuszczalnych w wodzie jest prekursorami koenzymów lub wchodzi w ich skład, np. witaminy B1, B2, B6, B7, B9 i B12[18].
Dinukleotyd nikotynoamidoadeninowy (NADH), pochodna witaminy B3 (niacyny), jest ważnym koenzymem pełniącym funkcję akceptora wodoru. Setki różnych typów dehydrogenaz odbierają elektrony substratom reakcji (utleniają i redukują NAD+ do NADH. Ta zredukowana postać koenzymu staje się następnie substratem przy tworzeniu różnych reduktaz, enzymów zajmujących się redukcją związków chemicznych[19]. Dinukleotyd nikotynoamidoadeninowy występuje w komórce w dwóch powiązanych ze sobą formach, NADH i NADPH. Formy NAD+/NADH są częściej wykorzystywane w reakcjach katabolicznych, podczas gdy NADP+/NADPH mają duże znaczenie dla przebiegu reakcji anabolicznych.
Związki nieorganiczne i kofaktory
Na około 99% masy przeciętnego ssaka składa się dziewięć pierwiastków: węgiel, wodór, tlen, azot, siarka, wapń, chlor, sód i potas[20]. Związki organiczne (białka, lipidy i węglowodany) skupiają większość węgla i azotu, podczas gdy największa część tlenu i wodoru zawarta jest w wodzie[20]. Wapń, chlor, sód i potas oraz pozostałe pierwiastki występujące w organizmach żywych są z kolei głównymi komponentami nieorganicznych związków chemicznych, z których część występuje w dużej ilości, natomiast inne potrzebne są w ilościach śladowych.
U większości organizmów główną część występujących w nich związków nieorganicznych stanowią jonowe elektrolity, takie jak jony sodu, potasu, wapnia, magnezu, chlorki, fosforany oraz wodorowęglany. Dokładne wartości stężeń poszczególnych jonów regulują mechanizmy ciśnienia osmotycznego i pH[21]. Związki nieorganiczne stanowią główny składnik struktur takich jak szkielet i muszle u tych organizmów, które je posiadają. Jony są również niezbędne do prawidłowego funkcjonowania komórek nerwowych i mięśni, jako że potencjał czynnościowy, pobudzający je do działania, powstaje podczas wymiany elektrolitów pomiędzy płynem pozakomórkowym a cytozolem[22]. Elektrolity dostają się do komórek i wydostają z nich poprzez kanały tworzone przez białka błony komórkowej zwane kanałami jonowymi. Przykładowo napięcie mięśniowe zależne jest od przepływu jonów wapnia, sodu i potasu przez owe kanały i tubule T (wpuklenia w błonie komórkowej włókien mięśniowych przyspieszające rozprzestrzenianie się impulsów elektrycznych)[23].
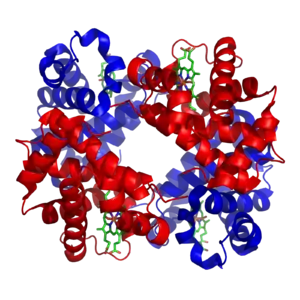
Metale przejściowe występują w organizmach w ilościach śladowych, z których najbardziej rozpowszechnione to cynk i żelazo[24][25]. Metale te są składnikami niektórych białek i kofaktorów, są również niezbędne dla funkcjonowania takich enzymów jak katalaza oraz białek transportujących tlen, na przykład hemoglobiny[26]. Kofaktory te związane są trwale z jednym typem białka, mimo że kofaktory enzymów mogą podczas katalizy ulegać modyfikacjom, po przeprowadzeniu reakcji zawsze wracają do postaci pierwotnej. Metale będące mikroelementami są przenoszone do komórek organizmu za pomocą specyficznych przenośników i wiązane z białkami przechowującymi je, np. ferrytyną czy metalotioneiną[27][28].
Katabolizm
Katabolizm stanowi grupa reakcji chemicznych, w których następuje rozkład lub utlenianie złożonych związków organicznych do związków prostszych z uwolnieniem energii. Ich wspólnym celem jest dostarczenie energii lub substratów niezbędnych do podtrzymania procesów życiowych organizmu. Szczegółowy charakter tych procesów jest różny dla poszczególnych grup organizmów. Jednak wszystkie te formy metabolizmu mają na celu utworzenie potencjału redoks pozwalającego na przenoszenie elektronów pomiędzy zredukowanymi cząstkami (takimi jak np. materia organiczna, amoniak, siarkowodór, jony żelaza) a akceptorami (na przykład tlenem, azotanami i siarczanami)[29]. W metabolizmie zwierząt reakcje te prowadzą do rozkładu cząstek organicznych do prostych związków, najczęściej dwutlenku węgla i wody, z uwolnieniem energii.
Najpowszechniejszy schemat reakcji katabolicznych w organizmach zwierząt można podzielić na trzy główne etapy. Podczas pierwszego z nich duże cząsteczki substancji organicznych – białek, polisacharydów czy lipidów są trawione w układzie pokarmowym do mniejszych cząsteczek. Następnie są one transportowane do komórek i rozkładane do jeszcze prostszych związków (najczęściej acetylo-CoA), podczas czego uwalniana jest energia. Wreszcie grupa acetylowa utleniana jest do dwutlenku węgla w cyklu Krebsa podczas którego energia przenoszona jest na NADH i GTP. Powstały NADH ulega utlenieniu w łańcuchu oddechowym, wyzwalając energię przechowywaną ostatecznie w ATP. Na tym etapie protony z NADH przenoszone są na tlen co prowadzi do wytworzenia drugiego produktu pełnego utlenienia związków organicznych – wody.
Trawienie
Makrocząsteczki takie jak skrobia, celuloza czy białka nie mogą być bezpośrednio wchłonięte przez komórki, muszą więc zostać wcześniej rozłożone do mniejszych cząsteczek. Głównymi grupami enzymów trawiennych są: proteazy rozkładające białka na aminokwasy, glukozydazy depolimeryzujące polisacharydy czy lipazy rozkładające lipidy do kwasów tłuszczowych. Mikroorganizmy wydzielają enzymy trawienne do swojego otoczenia[30][31], podczas gdy zwierzęta produkują je w odpowiednio wyspecjalizowanych komórkach w przewodzie pokarmowym[32]. Aminokwasy i cukry uwolnione przez te pozakomórkowe enzymy są następnie transportowane do wnętrza komórek za pomocą specjalnych białek w procesie transportu aktywnego[33][34].
Katabolizm związków organicznych

Wspólną cechą szlaków katabolicznych jest rozkładanie związków organicznych do mniejszych cząsteczek, w wyniku czego uwolniona zostaje energia w formie użytecznej dla komórki. Powstające małe cząsteczki chemiczne mogą być wykorzystane w komórce lub wydalane z niej. Katabolizm węglowodanów polega głównie na rozkładaniu ich na mniejsze cząsteczki. Są one transportowane do komórek wkrótce po rozłożeniu do monosacharydów[35]. Kolejnym etapem katabolicznego szlaku glukozy jest glikoliza, podczas której, z cukrów takich jak glukoza czy fruktoza powstaje kwas pirogronowy oraz energia wiązana w ATP[36]. Kwas pirogronowy jest elementem występującym w kilku szlakach metabolicznych, jednak zdecydowana większość jego cząsteczek jest przekształcana w acetylo-CoA i włączana w cykl kwasu cytrynowego. Mimo że podczas samego cyklu powstaje również kilka cząsteczek ATP, jego najważniejszym produktem jest NADH powstałe z NAD+ w chwili utleniania acetylo-CoA. Produktami końcowymi procesu utlenienia glukozy są cząsteczki CO
2, H
2O i energia. Alternatywną drogą rozkładu glukozy jest szlak pentozofosforanowy, podczas którego następuje redukcja koenzymu NADPH i produkcja cukrów z grupy pentoz takich jak ryboza – cukrowy komponent kwasu nukleinowego.
W warunkach beztlenowych pirogronian redukowany jest do kwasu mlekowego za pomocą enzymu dehydrogenazy mleczanowej, utleniającej ponownie NADH do NAD+, który może być ponownie użyty w glikolizie. Drugim sposobem odtworzenia NAD+ jest dekarboksylacja pirogronianu do aldehydu octowego, a następnie jego redukcja do etanolu przez dehydrogenazę alkoholową. Oba procesy nazywane są fermentacjami. W świecie mikroorganizmów zachodzi wiele innych fermentacji, poza opisanymi powyżej.
Katabolizm tłuszczów odbywa się poprzez proces hydrolizy, podczas którego uwalniane są kwasy tłuszczowe i glicerol. Glicerol przechodzi glikolizę, zaś kwasy tłuszczowe rozpadają się podczas beta-oksydacji z wytworzeniem acetylo-CoA, wchodzącego następnie w cykl kwasu cytrynowego. Utlenianie grama kwasów tłuszczowych wyzwala więcej energii niż utlenianie tej samej ilości glukozy, ponieważ węglowodany zawierają w swych strukturach więcej tlenu.
Aminokwasy mogą być użyte jako materiał do budowania białek i innych cząsteczek, lub też – po utlenieniu do mocznika, wody i dwutlenku węgla – jako źródło energii[37]. Proces oksydacji zaczyna się od usunięcia grupy aminowej podczas transaminacji. Wchodzi ona w cykl ornitynowy, pozostawiając szkielet węglowy w postaci ketokwasu. Niektóre z tych kwasów pełnią później różne role w cyklu kwasu cytrynowego, na przykład deaminują glutaminian – kwas α-ketoglutarowy[38]. Aminokwasy glukogenne mogą również przekształcić się w glukozę w procesie glukoneogenezy (patrz poniżej)[39].
Przemiana energii
Uporządkowanie struktur komórkowych i cząsteczek związków organicznych jest możliwe tylko dzięki stałemu dostarczaniu do komórki energii. Organizmy heterotroficzne zdolne są jedynie do pozyskiwania energii ze związków organicznych. Kluczowym elementem wytwarzania energii przydatnej dla komórki jest fosforylacja oksydacyjna. Organizmy określane nazwą fotoautotrofy zdolne są do wykorzystania energii światła i zamiany energii fal elektromagnetycznych na energię wiązań chemicznych. Niezbyt liczna grupa organizmów należących do prokariontów – chemoautotrofy posiada zdolność do wykorzystania energii pochodzącej z utleniania związków nieorganicznych w procesie chemosyntezy.
Fosforylacja oksydacyjna
W procesie fosforylacji oksydacyjnej, elektrony pobrane z cząsteczek związków organicznych w drodze m.in. cyklu kwasu cytrynowego przekazywane są na tlen, a uwolniona energia używana jest do tworzenia ATP. U eukariotów dzieje się to za pośrednictwem grupy białek występujących w błonie mitochondriów, zwanych łańcuchem oddechowym. U prokariotów białka te znajdują się w błonie wewnętrznej komórki[40]. Białka te używają energii wytworzonej podczas przemieszczania elektronów z cząsteczek zredukowanych (na przykład NADH) na cząsteczkę tlenu, aby przenosić protony poprzez wewnętrzną błonę komórkową[41]. Akceptorem elektronów u prokariontów mogą być, także inne związki niż tlen np. azotany[42], związki siarki[43].
Przeniesienie protonów z macierzy mitochondrialnej do przestrzeni międzybłonowej wytwarza różnicę stężeń i potencjałów pomiędzy obiema stronami błony i generuje potencjał elektrochemiczny[44]. Protony mogą powracać do macierzy mitochondrialnej poprzez kanał jonowy enzymu zwanego syntazą ATP. Przepływ ładunków dodatnich wywołuje rotację osi enzymu, dzięki czemu centrum aktywne syntazy zmienia kształt i fosforyluje ADP do ATP[17].
Energia ze związków nieorganicznych
Chemolitotrofia to rodzaj metabolizmu charakterystyczny dla niektórych organizmów prokariotycznych; pozyskują one energię z utleniania związków nieorganicznych. Mogą one używać wodoru[45], zredukowanych związków siarki (jonów S2-, siarkowodoru i tiosiarczanów S2O32-)[46], jonów żelaza(II) Fe2+[47] lub amoniaku[48] jako źródła potencjału redukcyjnego i czerpać energię z utleniania tych związków kosztem akceptorów takich jak tlen czy azotyny[49]. Te mikrobiologiczne procesy mają ogromne znaczenie w globalnych cyklach biogeochemicznych, takich jak acetogeneza, nitryfikacja i denitryfikacja; od ich przebiegu zależy także żyzność gleb[50][51].
Wiązanie energii słonecznej
U organizmów fotosyntetyzujących, takich jak rośliny i sinice, transfer elektronów nie jest efektem utleniania związków organicznych, lecz zachodzi dzięki pochłanianiu kwantów energii światła[52].
Energia słoneczna może być wiązana przez fotoautotrofy: rośliny, cyjanobakterie, bakterie purpurowe, zielone bakterie siarkowe i niektóre protisty. Proces ten jest często utożsamiany z wiązaniem dwutlenku węgla do związków organicznych w toku fotosyntezy. Jednak te dwa mechanizmy mogą u prokariotów funkcjonować niezależnie – przykładowo bakterie purpurowe i zielone siarkowe mogą używać światła jako źródła energii, przeprowadzając równocześnie proces wiązania węgla i fermentacji związków organicznych[53][54].
Wiązanie energii słonecznej to proces stosunkowo podobny do fosforylacji oksydacyjnej, jako że w jego toku powstaje gradient stężenia protonów, których przepływ przez syntazę ATP powoduje wytwarzanie adenozynotrójfosforanu[17]. Fotosyntetyczny łańcuch transportu elektronów napędzany jest przez światłoczułe kompleksy białkowe zwane fotosyntetycznym centrum reakcji. Kompleksy te dzielą się na dwa podstawowe rodzaje w zależności od długości fali przy której wykazują maksimum absorpcji, przy czym u większości fotosyntetyzujących bakterii wyróżnia się jeden typ centrum reakcji, a u roślin i cyjanobakterii – dwa[55].
U roślin fotosystem II wykorzystuje energię słoneczną do odbierania elektronów wodzie, co prowadzi do uwolnienia tlenu jako produktu ubocznego reakcji. Elektrony te następnie przechodzą do kompleksu cytochromów b6f, używającego ich energii do przenoszenia protonów przez błoną tylakoidową w chloroplaście[52]. Protony te wracając przez kanał jonowy syntazy ATP napędzają syntezę ATP, podobnie jak miało to miejsce w mitochondriach. Z kolei elektrony przechodzą do fotosystemu I, skąd po wybiciu kolejnym kwantem energii mogą być przeniesione na koenzym NADP+ (który jest niezbędny do przebiegu cyklu Calvina) lub wykorzystane do przenoszenia kolejnych protonów przez błonę tylakoidu[56].
Anabolizm
Anabolizm to grupa procesów metabolicznych, w których energia zużywana jest do syntezy złożonych cząsteczek. Te cząsteczki, stanowiące budulec wszystkich żywych komórek, są tworzone krok po kroku z prostych związków o stosunkowo niewielkich rozmiarach. Można wyróżnić trzy podstawowe etapy anabolizmu: pierwszy obejmuje produkcję aminokwasów, monosacharydów, izoprenoidów i nukleotydów, czyli podstawowych elementów biomolekuł. W drugim etapie cząsteczki te są aktywowane do form reaktywnych energią pochodzącą z ATP, zaś etap trzeci to łączenie wytworzonych cząsteczek w cząsteczki złożone – białka, polisacharydy, lipidy i kwasy nukleinowe.
Poszczególne organizmy różnią się liczbą typów wytwarzanych cząsteczek. Autotrofy, na przykład rośliny, budują w swych komórkach złożone cząsteczki z prostych cząstek, takich jak dwutlenek węgla i woda. Heterotrofy z kolei potrzebują do ich produkcji substancji bardziej złożonych – monosacharydów czy aminokwasów. Typ źródła energii może posłużyć za kryterium klasyfikacji organizmów: fotoautotrofy i fotoheterotrofy pozyskują energię ze światła słonecznego, a chemoautotrofy i chemoheterotrofy – z reakcji utleniania związków nieorganicznych.
Wiązanie węgla


Niemal wszystkie związki organiczne występujące obecnie na Ziemi powstały w procesie fotosyntezy. Fotosynteza to złożony proces podczas którego ze związków nieorganicznych, takich jak dwutlenek węgla (CO2) i woda, wytwarzane są związki organiczne. Do przeprowadzenia tego procesu wykorzystywana jest energia światła słonecznego, a produktem ubocznym jest zwykle tlen. W pierwszym etapie fotosyntezy wyprodukowane jest ATP i NADPH przez opisane powyżej fotosyntetyczne centra reakcji. Następnie oba związki zużywane są do syntezy aldehydu 3-fosfoglicerynowego przekształcanego stosunkowo prosto w glukozę. Reakcja wiązania węgla zachodzi dzięki obecności enzymu RuBisCO w cyklu Calvina-Bensona[57]. Rośliny przeprowadzają trzy typy fotosyntezy: C3, C4 i CAM. Zróżnicowanie to wynika z drogi, jaką CO2 dostaje się do cyklu Calvina: w fotosyntezie C3 jest on wiązany bezpośrednio, podczas gdy w C4 i CAM jest najpierw przekształcany do związków zawierających 4 atomy węgla; Dwa ostatnie typy fotosyntezy wykształciły się jako reakcja adaptacyjna na zmienne warunki oświetleniowe i wodne[58].
U fotosyntetyzujących organizmów prokariotycznych mechanizmy wiązania węgla są bardziej zróżnicowane. Proces ten może zachodzić w cyklu Calvina-Bensona, odwrotnym cyklu Krebsa[59] lub podczas karboksylacji acetylo-CoA[60][61]. Prokariotyczne chemoautotrofy również wiążą CO2 poprzez cykl Calvina-Bensona, do napędzania reakcji używają jednak energii pochodzącej z utleniania związków nieorganicznych[62].
Węglowodany i glikany
W anabolizmie węglowodanów proste kwasy organiczne mogą być przekształcane w monosacharydy takie jak glukoza, a następnie łączone w polisacharydy – na przykład skrobię. Synteza glukozy ze związków takich jak kwas pirogronowy, kwas mlekowy, glicerol, aldehyd 3-fosfoglicerynowy i aminokwasy zwana jest glukoneogenezą. Podczas tego procesu, w niektórych etapach powiązanego z glikolizą, kwas pirogronowy przekształcany jest do glukozo-6–fosforanu za pomocą szeregu reakcji[36]. Jakkolwiek, nie jest to zwykłe odwrócenie procesu glikolizy, ponieważ pewne reakcje katalizowane są przez enzymy nieglikolityczne. Jest to ważne, ponieważ tworzy rozdział między tworzeniem i rozpadem glukozy oraz nie dopuszcza do jednoczesnego przebiegu obu tych procesów[63][64].
Mimo że tłuszcze są typowym związkiem magazynującym energię, u kręgowców takich jak człowiek kwasy tłuszczowe nie mogą być przetworzone na glukozę w procesie glukogenezy, ponieważ organizmy te nie potrafią przekształcać acetylo-CoA do kwasu pirogronowego[65]. W związku z tym długotrwały głód zmusza organizmy kręgowców do produkcji ciał ketonowych zastępujących glukozę w organach takich jak mózg, które nie potrafią metabolizować kwasów tłuszczowych[66]. Inne organizmy, na przykład rośliny i bakterie, rozwiązały ten problem wprowadzając do swego metabolizmu cykl glioksylanowy. Omija on etap dekarboksylacji w cyklu Krebsa i transformuje acetyl-CoA do kwasu szczawiooctowego, który może być wykorzystany do produkcji glukozy[67][65].
Polisacharydy i glikany powstają w wyniku sekwencyjnego dołączania monosacharydów przez enzym – glikozylotransferazę – od reaktywnego donora (np. urydynodifosforanu) do akceptora grup hydroksylowych na powstającym polisacharydzie. Jako że każda z grup hydroksylowych pierścienia monosacharydu może być akceptorem, łańcuchy polisacharydów mają często rozgałęzioną strukturę[68]. Wyprodukowane polisacharydy mogą samodzielnie pełnić funkcje metaboliczne; mogą też być przekształcone do lipidów lub białek przez enzymy nazywane oligosacharyltransferazami[69][70].
Kwasy tłuszczowe, izoprenoidy i steroidy

Kwasy tłuszczowe powstają dzięki syntezie kwasów tłuszczowych, enzymowi polimeryzującemu i redukującemu jednostki acetylo-CoA. Ich łańcuchy acylowe są przedłużane w toku reakcji dołączania grup acylowych, redukowania ich do alkoholu, dehydratacji do grupy alkenowej i ponownej redukcji do alkanu. Enzymy biosyntezy kwasów tłuszczowych dzielą się na dwie grupy: u zwierząt i grzybów wszystkie te reakcje przeprowadzane są przez pojedyncze, multifunkcyjne białko typu I[71], podczas gdy w plastydach roślin i u bakterii poszczególne enzymy typu II przeprowadzają każdą reakcję z osobna[72][73].
Terpeny i izoprenoidy stanowią liczną grupę lipidów, w skład której wchodzą m.in. karotenoidy; tworzą one najliczniejszą klasę naturalnych produktów roślinnych[74]. Związki te powstają w procesie łączenia i modyfikowania jednostek izoprenowych dostarczanych przez pirofosforan izopentylu i pirofosforan dimetylallilu[75]. Owe pirofosforany mogą powstawać na różne sposoby. U zwierząt i archeobakterii są syntezowane w szlaku kwasu mewalonowego z cząsteczek acetylo-CoA[76], podczas gdy u roślin i bakterii szlak niemewalanowy używa jako substratów kwasu pirogronowego i aldehydu 3-fosfoglicerynowego[77][75]. Jedną z ważniejszych reakcji jakim ulegają owe donory izoprenu jest reakcja biosyntezy steroidów. Jednostki izoprenowe łączą się tu tworząc skwalen, a następnie są przekształcane w grupę pierścieni lanosterolu[78]. Ten może następnie być przekształcony w inne steroidy, np. cholesterol czy ergosterol[79][78].
Białka
Poszczególne organizmy mogą różnić się pod względem umiejętności syntezy 20 podstawowych aminokwasów. Większość bakterii i rośliny może syntezować wszystkie z nich, jednak ssaki posiadają zdolność syntezy jedynie 10[9]. Pozostałe 10 aminokwasów, w dodatku niezbędnych dla funkcjonowania organizmu, musi być dostarczane wraz z pożywieniem. Wszystkie one powstają dzięki procesom glikolizy, cyklu kwasu cytrynowego lub szlaku pentozofosforanowego. Azot dostarczany jest przez kwas glutaminowy i glutaminę. Synteza aminokwasów uzależniona jest od uformowania się odpowiednich cząsteczek alfa-ketokwasów, przechodzących w aminokwasy po transaminacji[80].
Aminokwasy przechodzą w białka w procesie łączenia ich wiązaniami peptydowymi w łańcuchy. Każde białko posiada unikalną sekwencję aminokwasów. Tak jak litery alfabetu mogą być łączone w niemal nieskończoną ilość kombinacji zwanych słowami, aminokwasy łączą się w sekwencje tworząc ogromne zróżnicowanie białek. Aminokwasy przed połączeniem muszą zostać aktywowane poprzez połączenie z cząsteczką tRNA za pomocą wiązania estrowego. Aminoacyl-tRNA powstaje w zależnej od ATP reakcji katalizowanej przez enzym – syntazę aminoacylu tRNA[81]. Ten aminoacyl-tRNA jest następnie włączany do powstającego łańcucha białkowego według informacji zawartej w mRNA[82].
Synteza i utylizacja nukleotydów
Nukleotydy powstają z aminokwasów, dwutlenku węgla i kwasu mrówkowego w procesach wymagających dużej ilości energii metabolicznej. Z tego powodu większość organizmów wykształciła mechanizmy powtórnego wykorzystywania nukleotydów[83][84][85]. Puryny syntezowane są tak jak nukleozydy. Zarówno adenina, jak i guanina powstają z pierwotnego nukleozydu inozyny, tworzonego z aminokwasów glicyny i glutaminy oraz kwasu asparaginowego i jonów mrówczanowych pochodzących z koenzymu tetrahydrofolianu. Piramidyny zaś syntezowane są z kwasu orotowego, który powstaje z glutaminy i kwasu asparaginowego[86].
Porównanie strategii metabolicznych
Organizmy żywe potrzebują do życia następujących substratów[87]:
- strukturalnych, z których są zbudowane, przede wszystkim pierwiastki C, H, O, N, S, z czego najważniejsze jest źródło węgla.
- donora elektronów, który dostarczy energii poprzez utlenienie się lub umożliwi zredukowanie utlenionych substratów (CO
2) w celu wytworzenia związków organicznych. - akceptora elektronów (utleniacza), który uwolni energię w reakcji z substratem energetycznym.
Ten sam związek może pełnić różne role. Wśród źródeł węgla można wyróżnić związki organiczne (z węglem na niskim stopniu utlenienia, u heterotrofów) bądź CO
2 wymagający energochłonnej redukcji (u autotrofów). Głównymi źródłami energii jest promieniowanie słoneczne i utlenianie związków chemicznych. Donory elektronów można podzielić na związki nieorganiczne i związki organiczne, a wśród akceptorów elektronów można dodatkowo wyróżnić tlen. U organizmów żywych można znaleźć większość z możliwych kombinacji szlaków metabolicznych, jednak największe znaczenie mają fotolitroautotrofy, chemolitoautotrofy oraz tlenowe i beztlenowe chemoorganoheterotrofy[87].
| Źródło energii | światło słoneczne | foto- | -trof | ||
| związki chemiczne | chemo- | ||||
| Źródło elektronów | związki organiczne | organo- | |||
| związki nieorganiczne | lito- | ||||
| Źródło węgla | związki organiczne | hetero- | |||
| związki nieorganiczne | auto- | ||||
| Fotosynteza | Fotosynteza anoksygeniczna | Chemosynteza | Oddychanie beztlenowe | Oddychanie tlenowe | Fermentacja | |
|---|---|---|---|---|---|---|
| Źródło węgla | asymilacja CO2[88] | asymilacja CO2 (poza halobakteriami); niektóre wykorzystują również inne związki organiczne (wówczas nie uznaje się je za autotrofy)[89] | asymilacja CO2[88] | związki organiczne | związki organiczne | związki organiczne |
| Źródło energii | energia świetlna[88] | energia świetlna[90] | utlenianie prostych związków nieorganicznych (siarczki, sole amonowe, azotyny, siarka rodzima, soli żelazawe, wodór) lub metanu[91] | utlenianie związków organicznych, wytworzenie gradientu elektrochemicznego[92], krótki łańcuch oddechowy[93] | utlenianie związków organicznych (glikoliza, cykl Krebsa); wytworzenie gradientu elektrochemicznego, łańcuch oddechowy | utlenianie związków organicznych przebiegające przez glikolizę (np. fermentacja alkoholowa) lub bez (np. heterofermentacja mlekowa)[94] |
| Źródło elektronów | woda (fotoliza wody przebiegająca z uwolnieniem tlenu)[90] | zredukowane związki nieorganiczne (zwłaszcza związki siarki, wodór: H2S, H2, S), rzadziej związki organiczne (jabłczan, mleczan u heliobakterii) lub zredukowane żelazo Fe2+ [90] |
proste związki nieorganiczne lub metan[91] | związki organiczne | związki organiczne | związki organiczne[95] (zwykle brak zewnętrznego akceptora elektronów, więc przenoszony jest on wprost na związek organiczny – substrat, aby odtworzyć NAD+)[96] |
| Akceptor elektronów | NADP+ (w fazie jasnej fotosyntezy)[97] | cykliczny transport elektronów (cykliczna fosforylacja)[97] | tlen (w przypadku organizmów tlenowych) lub utlenione substancje mineralne (siarczany, azotany, węglany)[91] | utlenione związki mineralne (np. siarczany, azotany), CO 2 lub rzadziej niektóre związki organiczne (np. fumaran)[98] |
tlen | związki organiczne; ten sam substrat daje jeden produkt utleniony, drugi zredukowany (reakcja dysmutacji); brak etapów pośrednich przenoszenia elektronów[96] |
| Przykłady organizmów | rośliny, sinice[90] | bakterie purpurowe siarkowe i bezsiarkowe, bakterie zielone[90] | bakterie nitryfikacyjne, bakterie wodorowe, bakterie żelazowe, bezbarwne bakterie siarkowe, metanotrofy[91] | bakterie denitryfikacyjne, bakterie desulfurykacyjne, metanogeny[98] | rośliny, grzyby, ssaki | drożdże Saccharomyces – fermentacja alkoholowa, bakterie kwasu mlekowego – fermentacja mlekowa[95] |
Ksenobiotyki i metabolizm reakcji redoks
Wszystkie organizmy są nieustannie wystawione na działanie związków chemicznych, których nie mogą użyć jako pożywienia i które mogłyby być szkodliwe w wypadku ich dostania się do komórek, ponieważ nie pełnią one żadnych funkcji metabolicznych. Te potencjalnie niebezpieczne substancje zwane są ksenobiotykami[99]. Ksenobiotyki takie jak leki, narkotyki, trucizny naturalne i antybiotyki są detoksyfikowane za pomocą określonych enzymów ksenobiotyczno-metabolicznych. U człowieka są to m.in. oksydaza cytochromu P450s[100], UDP–glukuronosyltransferaza[101] i S-transferaza glutationu[102]. Enzymy te działają w trzech etapach: utleniania ksenobiotyku (faza I), dołączania do jego cząsteczki grup hydrofilowych (faza II) i usuwania go z komórki wraz z wodą (u organizmów wielokomórkowych umożliwia to późniejsze strawienie), co stanowi fazę III. Reakcje te mają duże znaczenie dla sozologii, gdzie są podstawą biodegradacji substancji niebezpiecznych i bioremediacji skażonych gleb i wód[103]. Niezwykła różnorodność mikroorganizmów sprawia, że są one w stanie poradzić sobie z wszelkimi niemal typami ksenobiotyków[104].
Podobnym problemem dla organizmów tlenowych jest stres oksydacyjny[105]. Procesy takie jak fosforylacja oksydacyjna czy tworzenie wiązań disulfidowych podczas budowy białek powodują powstawanie reaktywnych form tlenu, np. nadtlenku wodoru[106]. Oksydanty te, powodujące zniszczenia w strukturach m.in. protein i kwasów nukleinowych, są neutralizowane przez przeciwutleniacze i enzymy: katalazy i peroksydazy[107][108].
Termodynamika w organizmach żywych
Reakcje zachodzące w organizmach żywych, podobnie jak wszystkie inne reakcje chemiczne, podlegają zasadom termodynamiki, opisującym przepływ energii cieplnej i pracy.
Zmiana entalpii swobodnej (ΔG) stanowi tę część energii całkowitej układu (a właściwie entalpii układu), którą można wykorzystać do wykonania pracy, jest energią użyteczną, odpowiednikiem potencjału chemicznego. Entropia to stopień nieuporządkowania układu) i zgodnie z drugą zasadą termodynamiki całkowita wartość entropii wykazuje tendencję wzrostową w procesach zachodzących samorzutnie, przybiera wartości maksymalne, gdy układ osiąga równowagę. Zależność między zmianą entalpii swobodnej (ΔG) w układzie a zmianą entropii (ΔS) w warunkach stałej temperatury i ciśnienia wyraża następujące równanie[109]:

gdzie: ΔH – zmiana entalpii (ciepło), T – temperatura bezwzględna
W warunkach, w jakich zachodzą reakcje biochemiczne, zmiana entalpii jest w przybliżeniu równa zmianie energii wewnętrznej reakcji (ΔE):

gdzie: ΔF – energia swobodna reakcji
Jeśli ΔG jest ujemna, to reakcja zachodzi samorzutnie, następuje utrata entalpii swobodnej (reakcja jest egzoenergetyczna), a przy dużych wartościach reakcja praktycznie przebiega tylko w jednym kierunku i jest zasadniczo nieodwracalna. Jeśli ΔG jest dodatnia, to reakcja zachodzi tylko wówczas, gdy dostarczona zostanie z zewnątrz energia (entalpia swobodna; reakcja jest endoenergetyczna). Przy dużych dodatnich wartościach można uznać, ze układ jest stabilny i wykazuje brak lub niewielkie skłonności do reagowania. Gdy ΔG równe jest zeru, wówczas układ znajduje się w stanie równowagi i nie zachodzi w nim żadna wypadkowa zmiana[109].
ATP jest głównym przenośnikiem energii w komórkach[1]. Energia swobodna uwolniona podczas hydrolizy ATP wykorzystywana jest do przeprowadzania reakcji wymagających jej dostarczenia (endoenergetycznych). Dzięki sprzężeniu takiej niekorzystnej termodynamicznie reakcji z reakcją wyzwalającą energię (rozkład ATP) następuje przesunięcie równowagi reakcji i możliwe zajście tej pierwszej[109].
Przykładowo pierwsza reakcja w glikolizie katalizowana przez enzym heksokinazę:
- glukoza + Pi → glukozo-6-fosforan + H2O (ΔGº = +13,8 kJ/mol)
- ATP → ADP + Pi (ΔGº = –30,5 kJ/mol)
po sprzężeniu:
- glukoza + ATP → glukozo-6-fosforan + ADP + H+ (ΔGº = –16,7 kJ/mol = –30,5 kJ/mol + 13,8 kJ/mol)
Ponieważ układ dąży do osiągnięcia stanu równowagi, energia swobodna reakcji ΔF (mogąca służyć do wykonania pracy) ma tendencję do obniżania się, a entropia – do zwiększania się. Im bardziej reakcja jest oddalona od stanu równowagi, tym mniej potencjału do wykonania pracy ulega utracie na rzecz wzrostu entropii. Dla metabolizmu komórkowego charakterystyczne jest, że stosunek ilości substratów i produktów pewnych reakcji nie znajduje się w stanie równowagi. O ile pewne reakcje w szlaku metabolicznym znajdują się jednak w stanie lub blisko stanu równowagi, to zawsze co najmniej jedna, a zwykle kilka znajduje się w stanie dalekim od równowagi, przez co są zasadniczo nieodwracalne i zachodzą w jednym kierunku. Podlegają one równocześnie mechanizmom regulacji poprzez stymulowanie lub hamowanie aktywności enzymów, które katalizują te reakcje[110]. Przykładowo gdyby zmierzyć ΔG w reakcjach glikolizy, okazałoby się, że wszystkie reakcje są bliskie stanu równowagi poza tymi, które katalizuje heksokinaza, fosfofruktokinaza I i kinaza pirogronianowa. Są one zasadniczo nieodwracalne, wiążą się z dużymi różnicami energii swobodnej, podczas gdy inne etapy charakteryzuje niewielki jej spadek[110].
Podstawowe zasady termodynamiki zostały sformułowane dla układów zamkniętych, nieożywionych[110] Mimo że wyjątkowa złożoność komórek zdaje się przeczyć drugiej zasadzie termodynamiki, faktem jest, że wszystkie żywe organizmy są tak naprawdę układami otwartymi wymieniającymi nieustannie materię i energię z otoczeniem. Nie pozostają one wprawdzie w stanie równowagi termodynamicznej, są jednak systemami dyssypatywnymi, utrzymującymi stan wysokiej złożoności dzięki zwiększaniu entropii swego otoczenia (np. przez wymianę substancji i energii z krwiobiegiem bądź podłożem hodowlanym)[111]. Jest to osiągane dzięki łączeniu spontanicznych (niewymagających doprowadzenia energii) reakcji katabolicznych z niespontanicznymi procesami anabolizmu. Druga zasada termodynamiki pozostaje spełniona, ponieważ wzrost stopnia uporządkowania wewnątrz organizmu, jest kompensowany obniżeniem stopnia uporządkowania w środowisku otaczającym. Tym samym entropia całego układu stale wzrasta. Można powiedzieć, że metabolizm utrzymuje porządek, tworząc nieporządek[112].
Stały przepływ tlenu oraz innych substancji do komórek i poza komórki pozwala na to, by jej metabolizm znajdował się w stanie ustalonym. Wówczas stężenia substratów i produktów różnych reakcji pozostają na względnie stałym poziomie, choć pojedyncze reakcje niekoniecznie znajdują się w stanie równowagi i niekoniecznie nie zmienia się poziom metabolitów. Komórki są zdolne do nieustannego dopasowywania stężenia kluczowych związków w zależności od zmieniających się warunków[110].
Regulacja i kontrola
Ponieważ środowisko większości organizmów podlega nieustannym zmianom, reakcje metaboliczne muszą być dokładnie regulowane dla utrzymania w komórce stanu stałości warunków zwanego homeostazą[113][114]. Regulacja metaboliczna pozwala również organizmom na odpowiadanie na bodźce zewnętrzne oraz warunkuje interakcję ze środowiskiem[115]. Dla zrozumienia mechanizmów regulacji szlaków metabolicznych niezbędne są definicje dwóch kluczowych pojęć. Po pierwsze, „regulacja” szlaku przez enzym to sposób, w jaki tempo jego przebiegu wzrasta lub spada w odpowiedzi na bodźce. Po drugie, „kontrola” sprawowana przez enzym to efekt, jaki zmiany te wywierają na ogólny przebieg szlaku[116]. Dla przykładu, enzym wykazujący zdolność znacznej modyfikacji aktywności nie będzie uwzględniony jako enzym kontrolujący dany szlak, jeśli ta modyfikacja aktywności wywierać będzie niewielki wpływ na ciąg procesów w tym szlaku[117].
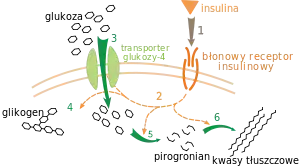
Regulacja metabolizmu zachodzi na wiele sposobów. Podstawowa regulacja szlaku metabolicznego polega na automatycznej odpowiedzi na zmianę stężenia substratów; przykładowo, zmniejszenie ilości produktów może – dla równowagi – przyspieszyć przebieg reakcji[116]. Często w ten sposób zachodzi regulacja allosteryczna aktywności poszczególnych enzymów szlaku[118]. Regulacja zewnętrzna wywołuje zmiany w metabolizmie komórki za pomocą sygnałów pochodzących z innych komórek; sygnały te mają zwykle postać rozpuszczalnych w wodzie substancji, takich jak hormony i czynniki wzrostu i są odbierane przez określone receptory na powierzchni komórki[119]. Są one następnie przekazywane do wnętrza komórki przez wewnętrzny łańcuch przekazywania sygnału, m.in. za pośrednictwem fosforylacji białek[120].
Przykładem bardzo dobrze poznanego mechanizmu regulacji zewnętrznej jest wpływ insuliny na metabolizm glukozy[121]. Insulina jest hormonem produkowanym w odpowiedzi na podwyższenie poziomu glukozy w organizmie. Łączenie się hormonu z receptorem insulinowym aktywuje grupę kinaz białkowych, które pobudzają komórki do pobierania glukozy z krwi i przekształcania jej w substancje zapasowe (na przykład kwasy tłuszczowe i glikogen[122]. Metabolizm glikogenu jest z kolei kontrolowany przez fosforylazę, enzym rozbijający glikogen, oraz tworzącą go syntazę glikogenu. Enzymy te są regulowane w sposób obustronny – fosforylacja dezaktywują syntazę glikogenu, aktywując jednocześnie fosforylazę. Insulina wywołuje syntezę glikogenu poprzez aktywację fosfatazy białkowej i hamowanie fosforylacji wymienionych enzymów[123].
Ewolucja

Najważniejsze z opisanych wyżej szlaków metabolicznych, na przykład glikoliza czy cykl kwasu cytrynowego, występują u organizmów wszystkich trzech domen i musiały pojawić się już u ich ostatniego wspólnego przodka[124][5]. Był to organizm prokariotyczny, zapewne metanogen o zewnętrznym metabolizmie aminokwasów, węglowodanów, nukleotydów i kwasów tłuszczowych[125][126]. Zatrzymanie dalszego rozwoju wymienionych szlaków podczas ewolucji może być wynikiem ich optymalnej wydajności, minimalnej złożoności i zaspokajaniu podstawowych potrzeb metabolicznych[6][7]. Przypuszcza się, że pierwsze szlaki metabolizmu oparte na działaniu enzymów mogły wchodzić w skład nukleotydowego metabolizmu puryn, wraz z wcześniejszym szlakami metabolicznymi tworzącymi część hipotetycznego świata RNA[127][128].
Powstało wiele hipotez tłumaczących mechanizm powstawania i ewolucji nowych szlaków metabolicznych. Zakładały one m.in. kolejne dodawanie nowych enzymów do krótkiego szlaku pierwotnego, duplikację i różnicowanie poszczególnych cykli, a także włączanie istniejących wcześniej enzymów do nowo powstających szlaków[129]. Wprawdzie względny udział tych mechanizmów w ewolucji nie został określony, badania genetyczne ujawniły jednak, że większość enzymów w obrębie danego szlaku ma zwykle wspólne pochodzenie, co wykazuje, że ich rozwój następował stopniowo i nowe funkcje powstawały na bazie już istniejących[130][131]. Inną możliwość stanowi istnienie uniwersalnych „modułów”, które – używane w różnych szlakach – wywierają podobny wpływ na różne substancje[132].
Ewolucja organizmów może również skutkować zanikaniem szlaków metabolicznych. Dla przykładu, u niektórych pasożytów pewne procesy metaboliczne przestają być niezbędne do życia, jako że gotowe aminokwasy, nukleotydy i węglowodany mogą być wchłaniane bezpośrednio od żywiciela[133][134]. Podobnie zredukowany metabolizm obserwowany jest u form endosymbiotycznych[135].
Metody badawcze
Metabolizm jest zazwyczaj badany za pomocą metod redukcjonistycznych, skupiających się na analizie poszczególnych szlaków metabolicznych i ich elementów. Jednym ze sposobów takiej analizy jest badanie drogi podanej substancji radioaktywnej w organizmie, od momentu jej podania do powstania końcowych produktów metabolizmu[137]. Enzymy katalizujące reakcje metaboliczne mogą zostać wyizolowane, co pozwala na zbadanie ich kinetyki i reakcji na inhibitory w warunkach in vitro. Jednocześnie możliwa jest identyfikacja mniejszych cząsteczek biorących udział w procesach metabolizmu; zbiór wszystkich takich substancji nazywany jest metabolomem. Ogólnie rzecz biorąc metoda ta jest skuteczna w przypadku badania struktury i funkcji prostych szlaków metabolicznych, zawodzi jednak przy procesach bardziej złożonych, np. całości metabolizmu komórki[138].
Sieci metaboliczne, ze względu na możliwą ilość interakcji pomiędzy substancjami oraz ilość samych substancji, są zazwyczaj bardzo skomplikowane. Stosowane techniki badawcze pozwalają jednak na rekonstruowanie całych sieci reakcji biochemicznych na podstawie informacji zawartych w genomie, co umożliwia budowanie holistycznych modeli matematycznych mogących wyjaśnić i przewidzieć ich przebieg[139]. Największą dokładność takich modeli uzyskuje się łącząc klasyczne metody badania metabolizmu z pochodzącą z badań nad proteomiką i sekwencją DNA wiedzą o ekspresji genu[140].
Informacje te znajdują zastosowanie przede wszystkim w inżynierii genetycznej. Organizmy takie jak drożdże, rośliny i bakterie są modyfikowane genetycznie w celu wykorzystania ich do produkcji różnych substancji: antybiotyków, insuliny czy witamin[141][142][143]. Modyfikacje genetyczne zazwyczaj mają na celu zmniejszenie kosztów i zwiększenie wydajności procesu wytwarzania produktu oraz redukcję ilości produktów ubocznych[144].
Historia badań nad metabolizmem

Termin „metabolizm” wywodzi się z greckiego słowa Μεταβολισμός – „Metabolismos” określającego zmianę, obalenie (np. rządu)[145]. Historia naukowych badań procesów metabolicznych obejmuje 400 lat, od początkowych badań nad zwierzętami do mikroskopowej analizy poszczególnych reakcji w nowoczesnej biochemii. Pierwsze eksperymenty mające na celu zbadanie ludzkiego metabolizmu wykonał i przedstawił w książce Ars de statica medecina Santorio Santorio[146]. Ważył on się przed i po jedzeniu, piciu, śnie, pracy, stosunku płciowym, poście i defekacji. Zauważył, że większość przyjmowanego pokarmu tracił poprzez – jak to określił – „nieświadomą perspirację”.
Podczas tych wczesnych badań nie utożsamiano jeszcze procesów życiowych z funkcjami metabolicznymi; za czynnik ożywiający uznawano wówczas nieznaną siłę życiową[147]. W XIX wieku, podczas badań nad fermentacją alkoholową przeprowadzaną przez drożdże, Louis Pasteur zauważył, że proces ten katalizowany jest przez substancje zawarte w komórkach drożdży, które nazwał „fermentami”. Z jego zapisków czytamy, że „fermentacja alkoholowa jest powiązana z procesami życiowymi i organizacją komórek drożdży, nie zaś z ich śmiercią czy rozkładem”[148]. Odkrycie to, które zbiegło się w czasie z pierwszą udaną syntezą związku organicznego (mocznika) z substancji nieorganicznych przeprowadzoną w 1828 r. przez Friedricha Wöhlera[149], udowodniło, że związki organiczne i reakcje zachodzące w komórkach nie różnią się ogólnym charakterem od innych zjawisk i substancji chemicznych.
Oddzielenie badań nad chemicznymi reakcjami metabolicznymi od nauki o biologii komórki nastąpiło wraz z odkryciem enzymów przez Eduarda Buchnera na początku XX wieku; chwila ta wyznacza umowny moment narodzin biochemii[150]. XX wiek przyniósł gwałtowny rozwój wiedzy z tej dziedziny, co w dużej mierze świat nauki zawdzięcza Hansowi Krebsowi[151]. Odkrył on istnienie cykli: ornitynowego, kwasu cytrynowego i glioksylanowego (2 ostatnie wraz z Hansem Kornbergiem)[152][153][67]. Ogromny wpływ na tempo rozwoju biochemii wywarły takie technologie, jak chromatografia, rentgenografia strukturalna, spektroskopia NMR, mikroskopia elektronowa i symulacje dynamiki molekularnej. Pozwoliły one na identyfikację i szczegółową analizę wielu cząsteczek i przebiegających w komórkach szlaków metabolicznych.
Zobacz też
Przypisy
- 1 2 3 Jeremy M. Berg, John L. Tymoczko, Lubert Stryer, Biochemia, Warszawa: Wydawnictwo Naukowe PWN, 2007, ISBN 978-83-01-14379-4.
- ↑ Kathleen M. Botham, Peter A. Mayes, Bioenergetyka: rola ATP, [w:] Robert Kincaid Murray, Daryl K. Granner, Victor W. Rodwell, Biochemia Harpera ilustrowana, wyd. 6, Warszawa: Wydawnictwo Lekarskie PZWL, 2008, s. 111–117, ISBN 978-83-200-3573-5.
- ↑ Cornelius G. Friedrich, Physiology and Genetics of Sulfur-oxidizing Bacteria, t. 39, Elsevier, 1997, s. 235–289, DOI: 10.1016/s0065-2911(08)60018-1, ISBN 978-0-12-027739-1, PMID: 9328649 (ang.).
- ↑ D.C. Dorman, Cytochrome Oxidase Inhibition Induced by Acute Hydrogen Sulfide Inhalation: Correlation with Tissue Sulfide Concentrations in the Rat Brain, Liver, Lung, and Nasal Epithelium, „Toxicological Sciences”, 65 (1), 2002, s. 18–25, DOI: 10.1093/toxsci/65.1.18 [dostęp 2023-11-26] (ang.).
- 1 2 Eric Smith, Harold J. Morowitz, Universality in intermediary metabolism, „Proceedings of the National Academy of Sciences of the United States of America”, 101 (36), 2004, s. 13168–13173, DOI: 10.1073/pnas.0404922101, PMID: 15340153, PMCID: PMC516543 [dostęp 2023-11-26] (ang.).
- 1 2 O. Ebenhöh, R. Heinrich, Evolutionary optimization of metabolic pathways. Theoretical reconstruction of the stoichiometry of ATP and NADH producing systems, „Bulletin of Mathematical Biology”, 63 (1), 2001, s. 21–55, DOI: 10.1006/bulm.2000.0197, PMID: 11146883 [dostęp 2023-11-26] (ang.).
- 1 2 E. Meléndez-Hevia, T.G. Waddell, M. Cascante, The puzzle of the Krebs citric acid cycle: assembling the pieces of chemically feasible reactions, and opportunism in the design of metabolic pathways during evolution, „Journal of Molecular Evolution”, 43 (3), 1996, s. 293–303, DOI: 10.1007/BF02338838, PMID: 8703096 [dostęp 2023-11-26] (ang.).
- ↑ Katharine A. Michie, Jan Löwe, Dynamic filaments of the bacterial cytoskeleton, „Annual Review of Biochemistry”, 75, 2006, s. 467–492, DOI: 10.1146/annurev.biochem.75.103004.142452, PMID: 16756499 [dostęp 2023-11-26] (ang.).
- 1 2 3 4 5 David L. Nelson, Michael M. Cox: Lehninger Principles of Biochemistry. W. H. Freeman and company, 2005, s. 841. ISBN 0-7167-4339-6. (ang.).
- ↑ Eoin Fahy i inni, A comprehensive classification system for lipids, „Journal of Lipid Research”, 46 (5), 2005, s. 839–861, DOI: 10.1194/jlr.E400004-JLR200, PMID: 15722563 [dostęp 2023-11-26] (ang.).
- ↑ Nomenclature of Lipids. IUPAC-IUB Commission on Biochemical Nomenclature (CBN). (ang.).
- ↑ F.G. Hegardt, Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis, „The Biochemical Journal”, 338 (Pt 3), 1999, s. 569–582, PMID: 10051425, PMCID: PMC1220089 [dostęp 2023-11-27] (ang.).
- ↑ F.G. Hegardt, Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis, „The Biochemical Journal”, 338 ( Pt 3) (Pt 3), 1999, s. 569–582, PMID: 10051425, PMCID: PMC1220089 [dostęp 2023-11-26] (ang.).
- ↑ Saleta Sierra, Bernd Kupfer, Rolf Kaiser, Basics of the virology of HIV-1 and its replication, „Journal of Clinical Virology: The Official Publication of the Pan American Society for Clinical Virology”, 34 (4), 2005, s. 233–244, DOI: 10.1016/j.jcv.2005.09.004, PMID: 16198625 [dostęp 2023-11-26] (ang.).
- 1 2 M.J. Wimmer, I.A. Rose, Mechanisms of enzyme-catalyzed group transfer reactions, „Annual Review of Biochemistry”, 47, 1978, s. 1031–1078, DOI: 10.1146/annurev.bi.47.070178.005123, PMID: 354490 [dostęp 2023-11-26] (ang.).
- ↑ P. Mitchell, The Ninth Sir Hans Krebs Lecture. Compartmentation and communication in living systems. Ligand conduction: a general catalytic principle in chemical, osmotic and chemiosmotic reaction systems, „European Journal of Biochemistry”, 95 (1), 1979, s. 1–20, DOI: 10.1111/j.1432-1033.1979.tb12934.x, PMID: 378655 [dostęp 2023-11-26] (ang.).
- 1 2 3 4 Peter Dimroth, Christoph von Ballmoos, Thomas Meier, Catalytic and mechanical cycles in F-ATP synthases. Fourth in the Cycles Review Series, „EMBO reports”, 7 (3), 2006, s. 276–282, DOI: 10.1038/sj.embor.7400646, PMID: 16607397, PMCID: PMC1456893 [dostęp 2023-11-26] (ang.).
- ↑ Większość rozpuszczalnych w wodzie witamin wchodzi w skład koenzymów, [w:] Lubert Stryer, Biochemia, wyd. 2, Warszawa: PWN, 2003, s. 482–483, ISBN 83-01-13978-1.
- ↑ Nadine Pollak, Christian Dölle, Mathias Ziegler, The power to reduce: pyridine nucleotides--small molecules with a multitude of functions, „The Biochemical Journal”, 402 (2), 2007, s. 205–218, DOI: 10.1042/BJ20061638, PMID: 17295611, PMCID: PMC1798440 [dostęp 2023-11-26] (ang.).
- 1 2 S.B. Heymsfield i inni, Chemical and elemental analysis of humans in vivo using improved body composition models, „The American Journal of Physiology”, 261 (2 Pt 1), 1991, E190–198, DOI: 10.1152/ajpendo.1991.261.2.E190, PMID: 1872381 [dostęp 2023-11-26] (ang.).
- ↑ H Sychrová, Yeast as a model organism to study transport and homeostasis of alkali metal cations, „Physiological Research”, 2004, S91–S98, DOI: 10.33549/physiolres.930000.53.S91, PMID: 15119939 [dostęp 2023-11-26] (ang.).
- ↑ I.B. Levitan, Modulation of ion channels in neurons and other cells, „Annual Review of Neuroscience”, 11, 1988, s. 119–136, DOI: 10.1146/annurev.ne.11.030188.001003, PMID: 2452594 [dostęp 2023-11-26] (ang.).
- ↑ A.F. Dulhunty, Excitation-contraction coupling from the 1950s into the new millennium, „Clinical and Experimental Pharmacology & Physiology”, 33 (9), 2006, s. 763–772, DOI: 10.1111/j.1440-1681.2006.04441.x, PMID: 16922804 [dostęp 2023-11-26] (ang.).
- ↑ D.C. Mahan, R.G. Shields, Macro- and micromineral composition of pigs from birth to 145 kilograms of body weight, „Journal of Animal Science”, 76 (2), 1998, s. 506–512, DOI: 10.2527/1998.762506x, PMID: 9498359 [dostęp 2023-11-26] (ang.).
- ↑ Søren Husted i inni, Elemental fingerprint analysis of barley ( Hordeum vulgare ) using inductively coupled plasma mass spectrometry, isotope-ratio mass spectrometry, and multivariate statistics, „Analytical and Bioanalytical Chemistry”, 378 (1), 2004, s. 171–182, DOI: 10.1007/s00216-003-2219-0, PMID: 14551660 [dostęp 2023-11-26] (ang.).
- ↑ Lydia A. Finney, Thomas V. O'Halloran, Transition metal speciation in the cell: insights from the chemistry of metal ion receptors, „Science”, 300 (5621), 2003, s. 931–936, DOI: 10.1126/science.1085049, PMID: 12738850 [dostęp 2023-11-26] (ang.).
- ↑ Robert J. Cousins, Juan P. Liuzzi, Louis A. Lichten, Mammalian zinc transport, trafficking, and signals, „Journal of Biological Chemistry”, 281 (34), 2006, s. 24085–24089, DOI: 10.1074/jbc.R600011200, PMID: 16793761 [dostęp 2023-11-26] (ang.).
- ↑ Louise L. Dunn, Yohan Suryo Rahmanto, Des R. Richardson, Iron uptake and metabolism in the new millennium, „Trends in Cell Biology”, 17 (2), 2007, s. 93–100, DOI: 10.1016/j.tcb.2006.12.003, PMID: 17194590 [dostęp 2023-11-26] (ang.).
- ↑ K.H. Nealson, P.G. Conrad, Life: past, present and future, „Philosophical Transactions of the Royal Society of London. Series B. Biological Sciences”, 354 (1392), 1999, s. 1923–1939, DOI: 10.1098/rstb.1999.0532, PMID: 10670014, PMCID: PMC1692713 [dostęp 2023-11-27] (ang.).
- ↑ C.C. Häse, R.A. Finkelstein, Bacterial extracellular zinc-containing metalloproteases, „Microbiological Reviews”, 57 (4), 1993, s. 823–837, DOI: 10.1128/mr.57.4.823-837.1993, PMID: 8302217, PMCID: PMC372940 [dostęp 2023-11-27] (ang.).
- ↑ R. Gupta, N. Gupta, P. Rathi, Bacterial lipases: an overview of production, purification and biochemical properties, „Applied Microbiology and Biotechnology”, 64 (6), 2004, s. 763–781, DOI: 10.1007/s00253-004-1568-8, PMID: 14966663 [dostęp 2023-11-27] (ang.).
- ↑ T. Hoyle, The digestive system: linking theory and practice, „British Journal of Nursing”, 6 (22), 1998, s. 1285–1291, DOI: 10.12968/bjon.1997.6.22.1285, PMID: 9470654 [dostęp 2023-11-27] (ang.).
- ↑ W.W. Souba, A.J. Pacitti, How amino acids get into cells: mechanisms, models, menus, and mediators, „JPEN. Journal of parenteral and enteral nutrition”, 16 (6), 1992, s. 569–578, DOI: 10.1177/0148607192016006569, PMID: 1494216 [dostęp 2023-11-27] (ang.).
- ↑ M.P. Barrett, A.R. Walmsley, G.W. Gould, Structure and function of facilitative sugar transporters, „Current Opinion in Cell Biology”, 11 (4), 1999, s. 496–502, DOI: 10.1016/s0955-0674(99)80072-6, PMID: 10449337 [dostęp 2023-11-27] (ang.).
- ↑ G.I. Bell i inni, Structure and function of mammalian facilitative sugar transporters, „Journal of Biological Chemistry”, 268 (26), 1993, s. 19161–19164, DOI: 10.1016/S0021-9258(19)36489-0, PMID: 8366068 [dostęp 2023-11-27] (ang.).
- 1 2 Clara Bouché i inni, The cellular fate of glucose and its relevance in type 2 diabetes, „Endocrine Reviews”, 25 (5), 2004, s. 807–830, DOI: 10.1210/er.2003-0026, PMID: 15466941 [dostęp 2023-11-27] (ang.).
- ↑ W. Sakami, H. Harrington, Amino acid metabolism, „Annual Review of Biochemistry”, 32, 1963, s. 355–398, DOI: 10.1146/annurev.bi.32.070163.002035, PMID: 14144484 [dostęp 2023-11-27] (ang.).
- ↑ J.T. Brosnan, Glutamate, at the interface between amino acid and carbohydrate metabolism, „The Journal of Nutrition”, 130 (4S Suppl), 2000, 988S–90S, DOI: 10.1093/jn/130.4.988S, PMID: 10736367 [dostęp 2023-11-27] (ang.).
- ↑ V.R. Young, A.M. Ajami, Glutamine: the emperor or his clothes?, „The Journal of Nutrition”, 131 (9 Suppl), 2001, 2449S-2459S, DOI: 10.1093/jn/131.9.2449S, PMID: 11533293 [dostęp 2023-11-27] (ang.).
- ↑ Jonathan P. Hosler, Shelagh Ferguson-Miller, Denise A. Mills, Energy transduction: proton transfer through the respiratory complexes, „Annual Review of Biochemistry”, 75, 2006, s. 165–187, DOI: 10.1146/annurev.biochem.75.062003.101730, PMID: 16756489, PMCID: PMC2659341 [dostęp 2023-11-27] (ang.).
- ↑ B.E. Schultz, S.I. Chan, Structures and proton-pumping strategies of mitochondrial respiratory enzymes, „Annual Review of Biophysics and Biomolecular Structure”, 30, 2001, s. 23–65, DOI: 10.1146/annurev.biophys.30.1.23, PMID: 11340051 [dostęp 2023-11-27] (ang.).
- ↑ Felipe Cava, Olga Zafra, José Berenguer, A cytochrome c containing nitrate reductase plays a role in electron transport for denitrification in Thermus thermophilus without involvement of the bc respiratory complex, „Molecular Microbiology”, 70 (2), 2008, s. 507–518, DOI: 10.1111/j.1365-2958.2008.06429.x, PMID: 18761683 [dostęp 2023-11-27] (ang.).
- ↑ Melike Balk i inni, Desulfatirhabdium butyrativorans gen. nov., sp. nov., a butyrate-oxidizing, sulfate-reducing bacterium isolated from an anaerobic bioreactor, „International Journal of Systematic and Evolutionary Microbiology”, 58 (Pt 1), 2008, s. 110–115, DOI: 10.1099/ijs.0.65396-0, PMID: 18175693 [dostęp 2023-11-27] (ang.).
- ↑ Roderick A. Capaldi, Robert Aggeler, Mechanism of the F(1)F(0)-type ATP synthase, a biological rotary motor, „Trends in Biochemical Sciences”, 27 (3), 2002, s. 154–160, DOI: 10.1016/s0968-0004(01)02051-5, PMID: 11893513 [dostęp 2023-11-27] (ang.).
- ↑ B. Friedrich, E. Schwartz, Molecular biology of hydrogen utilization in aerobic chemolithotrophs, „Annual Review of Microbiology”, 47, 1993, s. 351–383, DOI: 10.1146/annurev.mi.47.100193.002031, PMID: 8257102 [dostęp 2023-11-27] (ang.).
- ↑ C.G. Friedrich, Physiology and genetics of sulfur-oxidizing bacteria, „Advances in Microbial Physiology”, 39, 1998, s. 235–289, DOI: 10.1016/s0065-2911(08)60018-1, PMID: 9328649 [dostęp 2023-11-27] (ang.).
- ↑ Karrie A. Weber, Laurie A. Achenbach, John D. Coates, Microorganisms pumping iron: anaerobic microbial iron oxidation and reduction, „Nature Reviews. Microbiology”, 4 (10), 2006, s. 752–764, DOI: 10.1038/nrmicro1490, PMID: 16980937 [dostęp 2023-11-27] (ang.).
- ↑ M.S. Jetten i inni, The anaerobic oxidation of ammonium, „FEMS microbiology reviews”, 22 (5), 1998, s. 421–437, DOI: 10.1111/j.1574-6976.1998.tb00379.x, PMID: 9990725 [dostęp 2023-11-27] (ang.).
- ↑ Jörg Simon, Enzymology and bioenergetics of respiratory nitrite ammonification, „FEMS microbiology reviews”, 26 (3), 2002, s. 285–309, DOI: 10.1111/j.1574-6976.2002.tb00616.x, PMID: 12165429 [dostęp 2023-11-27] (ang.).
- ↑ R. Conrad, Soil microorganisms as controllers of atmospheric trace gases (H2, CO, CH4, OCS, N2O, and NO), „Microbiological Reviews”, 60 (4), 1996, s. 609–640, DOI: 10.1128/mr.60.4.609-640.1996, PMID: 8987358, PMCID: PMC239458 [dostęp 2023-11-27] (ang.).
- ↑ José-Miguel Barea i inni, Microbial co-operation in the rhizosphere, „Journal of Experimental Botany”, 56 (417), 2005, s. 1761–1778, DOI: 10.1093/jxb/eri197, PMID: 15911555 [dostęp 2023-11-27] (ang.).
- 1 2 Nathan Nelson, Adam Ben-Shem, The complex architecture of oxygenic photosynthesis, „Nature Reviews. Molecular Cell Biology”, 5 (12), 2004, s. 971–982, DOI: 10.1038/nrm1525, PMID: 15573135 [dostęp 2023-11-27] (ang.).
- ↑ Marcel T.J. van der Meer i inni, Diel variations in carbon metabolism by green nonsulfur-like bacteria in alkaline siliceous hot spring microbial mats from Yellowstone National Park, „Applied and Environmental Microbiology”, 71 (7), 2005, s. 3978–3986, DOI: 10.1128/AEM.71.7.3978-3986.2005, PMID: 16000812, PMCID: PMC1168979 [dostęp 2023-11-27] (ang.).
- ↑ M.A. Tichi, F.R. Tabita, Interactive control of Rhodobacter capsulatus redox-balancing systems during phototrophic metabolism, „Journal of Bacteriology”, 183 (21), 2001, s. 6344–6354, DOI: 10.1128/JB.183.21.6344-6354.2001, PMID: 11591679, PMCID: PMC100130 [dostęp 2023-11-27] (ang.).
- ↑ J.P. Allen, J.C. Williams, Photosynthetic reaction centers, „FEBS letters”, 438 (1-2), 1998, s. 5–9, DOI: 10.1016/s0014-5793(98)01245-9, PMID: 9821949 [dostęp 2023-11-27] (ang.).
- ↑ Yuri Munekage i inni, Cyclic electron flow around photosystem I is essential for photosynthesis, „Nature”, 429 (6991), 2004, s. 579–582, DOI: 10.1038/nature02598, PMID: 15175756 [dostęp 2023-11-27] (ang.).
- ↑ H.M. Miziorko, G.H. Lorimer, Ribulose-1,5-bisphosphate carboxylase-oxygenase, „Annual Review of Biochemistry”, 52, 1983, s. 507–535, DOI: 10.1146/annurev.bi.52.070183.002451, PMID: 6351728 [dostęp 2023-11-27] (ang.).
- ↑ Antony N. Dodd i inni, Crassulacean acid metabolism: plastic, fantastic, „Journal of Experimental Botany”, 53 (369), 2002, s. 569–580, DOI: 10.1093/jexbot/53.369.569, PMID: 11886877 [dostęp 2023-11-27] (ang.).
- ↑ Michael Hügler i inni, Evidence for autotrophic CO2 fixation via the reductive tricarboxylic acid cycle by members of the epsilon subdivision of proteobacteria, „Journal of Bacteriology”, 187 (9), 2005, s. 3020–3027, DOI: 10.1128/JB.187.9.3020-3027.2005, PMID: 15838028, PMCID: PMC1082812 [dostęp 2023-11-27] (ang.).
- ↑ G. Strauss, G. Fuchs, Enzymes of a novel autotrophic CO2 fixation pathway in the phototrophic bacterium Chloroflexus aurantiacus, the 3-hydroxypropionate cycle, „European Journal of Biochemistry”, 215 (3), 1993, s. 633–643, DOI: 10.1111/j.1432-1033.1993.tb18074.x, PMID: 8354269 [dostęp 2023-11-27] (ang.).
- ↑ H.G. Wood, Life with CO or CO2 and H2 as a source of carbon and energy, „FASEB journal”, 5 (2), 1991, s. 156–163, DOI: 10.1096/fasebj.5.2.1900793, PMID: 1900793 [dostęp 2023-11-27] (ang.).
- ↑ J.M. Shively, G. van Keulen, W.G. Meijer, Something from almost nothing: carbon dioxide fixation in chemoautotrophs, „Annual Review of Microbiology”, 52, 1998, s. 191–230, DOI: 10.1146/annurev.micro.52.1.191, PMID: 9891798 [dostęp 2023-11-27] (ang.).
- ↑ A. Boiteux, B. Hess, Design of glycolysis, „Philosophical Transactions of the Royal Society of London. Series B. Biological Sciences”, 293 (1063), 1981, s. 5–22, DOI: 10.1098/rstb.1981.0056, PMID: 6115423 [dostęp 2023-11-27] (ang.).
- ↑ S.J. Pilkis, M. R. el-Maghrabi, T.H. Claus, Fructose-2,6-bisphosphate in control of hepatic gluconeogenesis. From metabolites to molecular genetics, „Diabetes Care”, 13 (6), 1990, s. 582–599, DOI: 10.2337/diacare.13.6.582, PMID: 2162755 [dostęp 2023-11-27] (ang.).
- 1 2 Scott A. Ensign, Revisiting the glyoxylate cycle: alternate pathways for microbial acetate assimilation, „Molecular Microbiology”, 61 (2), 2006, s. 274–276, DOI: 10.1111/j.1365-2958.2006.05247.x, PMID: 16856935 [dostęp 2023-11-27] (ang.).
- ↑ Patrick F. Finn, J. Fred Dice, Proteolytic and lipolytic responses to starvation, „Nutrition”, 22 (7-8), 2006, s. 830–844, DOI: 10.1016/j.nut.2006.04.008, PMID: 16815497 [dostęp 2023-11-27] (ang.).
- 1 2 H.L. Kornberg, H.A. Krebs, Synthesis of cell constituents from C2-units by a modified tricarboxylic acid cycle, „Nature”, 179 (4568), 1957, s. 988–991, DOI: 10.1038/179988a0, PMID: 13430766 [dostęp 2023-11-27] (ang.).
- ↑ T.W. Rademacher, R.B. Parekh, R.A. Dwek, Glycobiology, „Annual Review of Biochemistry”, 57, 1988, s. 785–838, DOI: 10.1146/annurev.bi.57.070188.004033, PMID: 3052290 [dostęp 2023-11-27] (ang.).
- ↑ G. Opdenakker i inni, Concepts and principles of glycobiology, „FASEB journal”, 7 (14), 1993, s. 1330–1337, DOI: 10.1096/fasebj.7.14.8224606, PMID: 8224606 [dostęp 2023-11-27] (ang.).
- ↑ M.J. McConville, A.K. Menon, Recent developments in the cell biology and biochemistry of glycosylphosphatidylinositol lipids (review), „Molecular Membrane Biology”, 17 (1), 2000, s. 1–16, DOI: 10.1080/096876800294443, PMID: 10824734 [dostęp 2023-11-27] (ang.).
- ↑ Subrahmanyam S. Chirala, Salih J. Wakil, Structure and function of animal fatty acid synthase, „Lipids”, 39 (11), 2004, s. 1045–1053, DOI: 10.1007/s11745-004-1329-9, PMID: 15726818 [dostęp 2023-11-27] (ang.).
- ↑ Stephen W. White i inni, The structural biology of type II fatty acid biosynthesis, „Annual Review of Biochemistry”, 74, 2005, s. 791–831, DOI: 10.1146/annurev.biochem.74.082803.133524, PMID: 15952903 [dostęp 2023-11-27] (ang.).
- ↑ John B. Ohlrogge, Jan G. Jaworski, REGULATION OF FATTY ACID SYNTHESIS, „Annual Review of Plant Physiology and Plant Molecular Biology”, 48, 1997, s. 109–136, DOI: 10.1146/annurev.arplant.48.1.109, PMID: 15012259 [dostęp 2023-11-27] (ang.).
- ↑ Vinod Shanker Dubey, Ritu Bhalla, Rajesh Luthra, An overview of the non-mevalonate pathway for terpenoid biosynthesis in plants, „Journal of Biosciences”, 28 (5), 2003, s. 637–646, DOI: 10.1007/BF02703339, PMID: 14517367 [dostęp 2023-11-27] (ang.).
- 1 2 Tomohisa Kuzuyama, Haruo Seto, Diversity of the biosynthesis of the isoprene units, „Natural Product Reports”, 20 (2), 2003, s. 171–183, DOI: 10.1039/b109860h, PMID: 12735695 [dostęp 2023-11-27] (ang.).
- ↑ Laura L. Grochowski, Huimin Xu, Robert H. White, Methanocaldococcus jannaschii uses a modified mevalonate pathway for biosynthesis of isopentenyl diphosphate, „Journal of Bacteriology”, 188 (9), 2006, s. 3192–3198, DOI: 10.1128/JB.188.9.3192-3198.2006, PMID: 16621811, PMCID: PMC1447442 [dostęp 2023-11-27] (ang.).
- ↑ Hartmut K. Lichtenthaler, The 1-Ddeoxy-D-xylulose-5–phosphate pathway of isoprenoid biosynthesis in plants, „Annual Review of Plant Physiology and Plant Molecular Biology”, 50, 1999, s. 47–65, DOI: 10.1146/annurev.arplant.50.1.47, PMID: 15012203 [dostęp 2023-11-27] (ang.).
- 1 2 G.J. Schroepfer, Sterol biosynthesis, „Annual Review of Biochemistry”, 50, 1981, s. 585–621, DOI: 10.1146/annurev.bi.50.070181.003101, PMID: 7023367 [dostęp 2023-11-27] (ang.).
- ↑ N.D. Lees i inni, Cloning of the late genes in the ergosterol biosynthetic pathway of Saccharomyces cerevisiae--a review, „Lipids”, 30 (3), 1995, s. 221–226, DOI: 10.1007/BF02537824, PMID: 7791529 [dostęp 2023-11-27] (ang.).
- ↑ Arthur C. Guyton, John E. Hall, Textbook of Medical Physiology, Elsevier, 2006, s. 855–6, ISBN 0-7216-0240-1.
- ↑ M. Ibba, D. Söll, The renaissance of aminoacyl-tRNA synthesis, „EMBO reports”, 2 (5), 2001, s. 382–387, DOI: 10.1093/embo-reports/kve095, PMID: 11375928, PMCID: PMC1083889 [dostęp 2023-11-27] (ang.).
- ↑ P. Lengyel, D. Söll, Mechanism of protein biosynthesis, „Bacteriological Reviews”, 33 (2), 1969, s. 264–301, DOI: 10.1128/br.33.2.264-301.1969, PMID: 4896351, PMCID: PMC378322 [dostęp 2023-11-27] (ang.).
- ↑ F.B. Rudolph, The biochemistry and physiology of nucleotides, „The Journal of Nutrition”, 124 (1 Suppl), 1994, 124S–127S, DOI: 10.1093/jn/124.suppl_1.124S, PMID: 8283301 [dostęp 2023-11-27] (ang.).
- ↑ Rita Zrenner i inni, Pyrimidine and purine biosynthesis and degradation in plants, „Annual Review of Plant Biology”, 57, 2006, s. 805–836, DOI: 10.1146/annurev.arplant.57.032905.105421, PMID: 16669783 [dostęp 2023-11-27] (ang.).
- ↑ Claudio Stasolla i inni, Purine and pyrimidine nucleotide metabolism in higher plants, „Journal of Plant Physiology”, 160 (11), 2003, s. 1271–1295, DOI: 10.1078/0176-1617-01169, PMID: 14658380 [dostęp 2023-11-27] (ang.).
- ↑ J.L. Smith, Enzymes of nucleotide synthesis, „Current Opinion in Structural Biology”, 5 (6), 1995, s. 752–757, DOI: 10.1016/0959-440x(95)80007-7, PMID: 8749362 [dostęp 2023-11-27] (ang.).
- 1 2 Weiner 2005 ↓, s. 84–86.
- 1 2 3 Kunicki-Goldfinger 1994 ↓, s. 178–179.
- ↑ Kunicki-Goldfinger 1994 ↓, s. 190–197.
- 1 2 3 4 5 Weiner 2005 ↓, s. 89–91.
- 1 2 3 4 Weiner 2005 ↓, s. 87–89.
- ↑ Prescott 2002 ↓, s. 190.
- ↑ Kunicki-Goldfinger 1994 ↓, s. 141–147.
- ↑ Kunicki-Goldfinger 1994 ↓, s. 158–166.
- 1 2 Weiner 2005 ↓, s. 95.
- 1 2 Kunicki-Goldfinger 1994 ↓, s. 147–148.
- 1 2 Stearns J.C., Surette M.G., Kaiser J.C: Microbiology for Dummies. Willey, 2015, s. 134. ISBN 978-1-118-87118-8.
- 1 2 Weiner 2005 ↓, s. 93–95.
- ↑ Bernard Testa, Stefanie D. Krämer, The biochemistry of drug metabolism--an introduction: part 1. Principles and overview, „Chemistry & Biodiversity”, 3 (10), 2006, s. 1053–1101, DOI: 10.1002/cbdv.200690111, PMID: 17193224 [dostęp 2023-11-27] (ang.).
- ↑ P.B. Danielson, The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans, „Current Drug Metabolism”, 3 (6), 2002, s. 561–597, DOI: 10.2174/1389200023337054, PMID: 12369887 [dostęp 2023-11-28] (ang.).
- ↑ C.D. King i inni, UDP-glucuronosyltransferases, „Current Drug Metabolism”, 1 (2), 2000, s. 143–161, DOI: 10.2174/1389200003339171, PMID: 11465080 [dostęp 2023-11-28] (ang.).
- ↑ D. Sheehan i inni, Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily, „The Biochemical Journal”, 360 (Pt 1), 2001, s. 1–16, DOI: 10.1042/0264-6021:3600001, PMID: 11695986, PMCID: PMC1222196 [dostęp 2023-11-28] (ang.).
- ↑ Teca Calcagno Galvão, William W. Mohn, Víctor de Lorenzo, Exploring the microbial biodegradation and biotransformation gene pool, „Trends in Biotechnology”, 23 (10), 2005, s. 497–506, DOI: 10.1016/j.tibtech.2005.08.002, PMID: 16125262 [dostęp 2023-11-28] (ang.).
- ↑ Dick B. Janssen i inni, Bacterial degradation of xenobiotic compounds: evolution and distribution of novel enzyme activities, „Environmental Microbiology”, 7 (12), 2005, s. 1868–1882, DOI: 10.1111/j.1462-2920.2005.00966.x, PMID: 16309386 [dostęp 2023-11-28] (ang.).
- ↑ K.J. Davies, Oxidative stress: the paradox of aerobic life, „Biochemical Society Symposium”, 61, 1995, s. 1–31, DOI: 10.1042/bss0610001, PMID: 8660387 [dostęp 2023-11-28] (ang.).
- ↑ Benjamin P. Tu, Jonathan S. Weissman, Oxidative protein folding in eukaryotes: mechanisms and consequences, „The Journal of Cell Biology”, 164 (3), 2004, s. 341–346, DOI: 10.1083/jcb.200311055, PMID: 14757749, PMCID: PMC2172237 [dostęp 2023-11-28] (ang.).
- ↑ H. Sies, Oxidative stress: oxidants and antioxidants, „Experimental Physiology”, 82 (2), 1997, s. 291–295, DOI: 10.1113/expphysiol.1997.sp004024, PMID: 9129943 [dostęp 2023-11-28] (ang.).
- ↑ Silvia Vertuani, Angela Angusti, Stefano Manfredini, The antioxidants and pro-antioxidants network: an overview, „Current Pharmaceutical Design”, 10 (14), 2004, s. 1677–1694, DOI: 10.2174/1381612043384655, PMID: 15134565 [dostęp 2023-11-28] (ang.).
- 1 2 3 Murray R. K., Granner D. K., Rodwell V. W.: Biochemia Harpera. Warszawa: Wydawnictwo Lekarskie PZWL, 2017, s. 111–117. ISBN 978-83-200-4554-3.
- 1 2 3 4 Karp G.: Cell and Molecular Biology. Wiley, 2010, s. 91–92, 108. ISBN 978-0-470-48337-4.
- ↑ U. von Stockar, J. Liu, Does microbial life always feed on negative entropy? Thermodynamic analysis of microbial growth, „Biochimica et Biophysica Acta”, 1412 (3), 1999, s. 191–211, DOI: 10.1016/s0005-2728(99)00065-1, PMID: 10482783 [dostęp 2023-11-28] (ang.).
- ↑ Y. Demirel, S.I. Sandler, Thermodynamics and bioenergetics, „Biophysical Chemistry”, 97 (2-3), 2002, s. 87–111, DOI: 10.1016/s0301-4622(02)00069-8, PMID: 12050002 [dostęp 2023-11-28] (ang.).
- ↑ Réka Albert, Scale-free networks in cell biology, „Journal of Cell Science”, 118 (Pt 21), 2005, s. 4947–4957, DOI: 10.1242/jcs.02714, PMID: 16254242 [dostęp 2023-11-28] (ang.).
- ↑ M.D. Brand, Regulation analysis of energy metabolism, „The Journal of Experimental Biology”, 200 (Pt 2), 1997, s. 193–202, DOI: 10.1242/jeb.200.2.193, PMID: 9050227 [dostęp 2023-11-28] (ang.).
- ↑ Orkun S. Soyer, Marcel Salathé, Sebastian Bonhoeffer, Signal transduction networks: topology, response and biochemical processes, „Journal of Theoretical Biology”, 238 (2), 2006, s. 416–425, DOI: 10.1016/j.jtbi.2005.05.030, PMID: 16045939 [dostęp 2023-11-28] (ang.).
- 1 2 M. Salter, R.G. Knowles, C.I. Pogson, Metabolic control, „Essays in Biochemistry”, 28, 1994, s. 1–12, PMID: 7925313 [dostęp 2023-11-28] (ang.).
- ↑ H.V. Westerhoff, A.K. Groen, R.J. Wanders, Modern theories of metabolic control and their applications (review), „Bioscience Reports”, 4 (1), 1984, s. 1–22, DOI: 10.1007/BF01120819, PMID: 6365197 [dostęp 2023-11-28] (ang.).
- ↑ D.A. Fell, S. Thomas, Physiological control of metabolic flux: the requirement for multisite modulation, „The Biochemical Journal”, 311 ( Pt 1) (Pt 1), 1995, s. 35–39, DOI: 10.1042/bj3110035, PMID: 7575476, PMCID: PMC1136115 [dostęp 2023-11-28] (ang.).
- ↑ Wayne A. Hendrickson, Transduction of biochemical signals across cell membranes, „Quarterly Reviews of Biophysics”, 38 (4), 2005, s. 321–330, DOI: 10.1017/S0033583506004136, PMID: 16600054 [dostęp 2023-11-28] (ang.).
- ↑ P. Cohen, The regulation of protein function by multisite phosphorylation--a 25 year update, „Trends in Biochemical Sciences”, 25 (12), 2000, s. 596–601, DOI: 10.1016/s0968-0004(00)01712-6, PMID: 11116185 [dostęp 2023-11-28] (ang.).
- ↑ G.E. Lienhard i inni, How cells absorb glucose, „Scientific American”, 266 (1), 1992, s. 86–91, DOI: 10.1038/scientificamerican0192-86, PMID: 1734513 [dostęp 2023-11-28] (ang.).
- ↑ Peter J. Roach, Glycogen and its metabolism, „Current Molecular Medicine”, 2 (2), 2002, s. 101–120, DOI: 10.2174/1566524024605761, PMID: 11949930 [dostęp 2023-11-28] (ang.).
- ↑ C.B. Newgard i inni, Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1, „Diabetes”, 49 (12), 2000, s. 1967–1977, DOI: 10.2337/diabetes.49.12.1967, PMID: 11117996 [dostęp 2023-11-28] (ang.).
- ↑ A.H. Romano, T. Conway, Evolution of carbohydrate metabolic pathways, „Research in Microbiology”, 147 (6-7), 1996, s. 448–455, DOI: 10.1016/0923-2508(96)83998-2, PMID: 9084754 [dostęp 2023-11-28] (ang.).}
- ↑ A.L. Koch, How did bacteria come to be?, „Advances in Microbial Physiology”, 40, 1998, s. 353–399, DOI: 10.1016/s0065-2911(08)60135-6, PMID: 9889982 [dostęp 2023-11-28] (ang.).
- ↑ C. Ouzounis, N. Kyrpides, The emergence of major cellular processes in evolution, „FEBS letters”, 390 (2), 1996, s. 119–123, DOI: 10.1016/0014-5793(96)00631-x, PMID: 8706840 [dostęp 2023-11-28] (ang.).
- ↑ Walter Gilbert, Origin of life: The RNA world, „Nature”, 319 (6055), 1986, s. 618, DOI: 10.1038/319618a0 [dostęp 2023-11-28] (ang.).
- ↑ Gustavo Caetano-Anollés, Hee Shin Kim, Jay E. Mittenthal, The origin of modern metabolic networks inferred from phylogenomic analysis of protein architecture, „Proceedings of the National Academy of Sciences of the United States of America”, 104 (22), 2007, s. 9358–9363, DOI: 10.1073/pnas.0701214104, PMID: 17517598, PMCID: PMC1890499 [dostęp 2023-11-28] (ang.).
- ↑ Steffen Schmidt i inni, Metabolites: a helping hand for pathway evolution?, „Trends in Biochemical Sciences”, 28 (6), 2003, s. 336–341, DOI: 10.1016/S0968-0004(03)00114-2, PMID: 12826406 [dostęp 2023-11-28] (ang.).
- ↑ Sara Light, Per Kraulis, Network analysis of metabolic enzyme evolution in Escherichia coli, „BMC bioinformatics”, 5, 2004, s. 15, DOI: 10.1186/1471-2105-5-15, PMID: 15113413, PMCID: PMC394313 [dostęp 2023-11-28] (ang.).
- ↑ Rui Alves, Raphael A.G. Chaleil, Michael J.E. Sternberg, Evolution of enzymes in metabolism: a network perspective, „Journal of Molecular Biology”, 320 (4), 2002, s. 751–770, DOI: 10.1016/s0022-2836(02)00546-6, PMID: 12095253 [dostęp 2023-11-28] (ang.).
- ↑ Victor Spirin i inni, A metabolic network in the evolutionary context: multiscale structure and modularity, „Proceedings of the National Academy of Sciences of the United States of America”, 103 (23), 2006, s. 8774–8779, DOI: 10.1073/pnas.0510258103, PMID: 16731630, PMCID: PMC1482654 [dostęp 2023-11-28] (ang.).
- ↑ Jeffrey G. Lawrence, Common themes in the genome strategies of pathogens, „Current Opinion in Genetics & Development”, 15 (6), 2005, s. 584–588, DOI: 10.1016/j.gde.2005.09.007, PMID: 16188434 [dostęp 2023-11-28] (ang.).
- ↑ Jennifer J. Wernegreen, For better or worse: genomic consequences of intracellular mutualism and parasitism, „Current Opinion in Genetics & Development”, 15 (6), 2005, s. 572–583, DOI: 10.1016/j.gde.2005.09.013, PMID: 16230003 [dostęp 2023-11-28] (ang.).
- ↑ Csaba Pál i inni, Chance and necessity in the evolution of minimal metabolic networks, „Nature”, 440 (7084), 2006, s. 667–670, DOI: 10.1038/nature04568, PMID: 16572170 [dostęp 2023-11-28] (ang.).
- ↑ Lieven Sterck i inni, How many genes are there in plants (... and why are they there)?, „Current Opinion in Plant Biology”, 10 (2), 2007, s. 199–203, DOI: 10.1016/j.pbi.2007.01.004, PMID: 17289424 [dostęp 2023-11-28] (ang.).
- ↑ M.J. Rennie, An introduction to the use of tracers in nutrition and metabolism, „The Proceedings of the Nutrition Society”, 58 (4), 1999, s. 935–944, DOI: 10.1017/s002966519900124x, PMID: 10817161 [dostęp 2023-11-28] (ang.).
- ↑ R.D. Phair, Development of kinetic models in the nonlinear world of molecular cell biology, „Metabolism: Clinical and Experimental”, 46 (12), 1997, s. 1489–1495, DOI: 10.1016/s0026-0495(97)90154-2, PMID: 9439549 [dostęp 2023-11-28] (ang.).
- ↑ Irina Borodina, Jens Nielsen, From genomes to in silico cells via metabolic networks, „Current Opinion in Biotechnology”, 16 (3), 2005, s. 350–355, DOI: 10.1016/j.copbio.2005.04.008, PMID: 15961036 [dostęp 2023-11-28] (ang.).
- ↑ Erwin P. Gianchandani, David L. Brautigan, Jason A. Papin, Systems analyses characterize integrated functions of biochemical networks, „Trends in Biochemical Sciences”, 31 (5), 2006, s. 284–291, DOI: 10.1016/j.tibs.2006.03.007, PMID: 16616498 [dostęp 2023-11-28] (ang.).
- ↑ Jette Thykaer, Jens Nielsen, Metabolic engineering of beta-lactam production, „Metabolic Engineering”, 5 (1), 2003, s. 56–69, DOI: 10.1016/s1096-7176(03)00003-x, PMID: 12749845 [dostęp 2023-11-28] (ang.).
- ↑ María González-Pajuelo i inni, Metabolic engineering of Clostridium acetobutylicum for the industrial production of 1,3-propanediol from glycerol, „Metabolic Engineering”, 7 (5-6), 2005, s. 329–336, DOI: 10.1016/j.ymben.2005.06.001, PMID: 16095939 [dostęp 2023-11-28] (ang.).
- ↑ Marco Krämer i inni, Metabolic engineering for microbial production of shikimic acid, „Metabolic Engineering”, 5 (4), 2003, s. 277–283, DOI: 10.1016/j.ymben.2003.09.001, PMID: 14642355 [dostęp 2023-11-28] (ang.).
- ↑ M. Koffas i inni, Metabolic engineering, „Annual Review of Biomedical Engineering”, 1, 1999, s. 535–557, DOI: 10.1146/annurev.bioeng.1.1.535, PMID: 11701499 [dostęp 2023-11-28] (ang.).
- ↑ Metabolism. The Online Etymology Dictionary. [dostęp 2007-02-20].
- ↑ G. Eknoyan, Santorio Sanctorius (1561–1636) - founding father of metabolic balance studies, „American Journal of Nephrology”, 19 (2), 1999, s. 226–233, DOI: 10.1159/000013455, PMID: 10213823 [dostęp 2023-11-28] (ang.).
- ↑ H.S. Williams, A History of Science: in Five Volumes. Volume IV: Modern Development of the Chemical and Biological Sciences, New York: Harper and Brothers, 1904 [zarchiwizowane 2006-08-30].
- ↑ K.L. Manchester, Louis Pasteur (1822–1895) – chance and the prepared mind, „Trends in Biotechnology”, 13 (12), 1995, s. 511–515, DOI: 10.1016/S0167-7799(00)89014-9, PMID: 8595136 [dostęp 2023-11-28] (ang.).
- ↑ E. Kinne-Saffran, R.K. Kinne, Vitalism and synthesis of urea. From Friedrich Wöhler to Hans A. Krebs, „American Journal of Nephrology”, 19 (2), 1999, s. 290–294, DOI: 10.1159/000013463, PMID: 10213830 [dostęp 2023-11-28] (ang.).
- ↑ Eduard Buchner, Cell-Free Fermentation. Nobel Lecture [online], NobelPrize.org [dostęp 2023-11-28] (ang.), The Nobel Prize in Chemistry 1907.
- ↑ H. Kornberg, Krebs and his trinity of cycles, „Nature Reviews. Molecular Cell Biology”, 1 (3), 2000, s. 225–228, DOI: 10.1038/35043073, PMID: 11252898 [dostęp 2023-11-28] (ang.).
- ↑ Hans Adolf Krebs, Kurt Henseleit, Untersuchungen uber die Harnstoffbildung im Tierkörper, „Hoppe-Seyler´s Zeitschrift für physiologische Chemie”, 210 (1-2), 1932, s. 33–66, DOI: 10.1515/bchm2.1932.210.1-2.33 [dostęp 2023-11-28] (niem.).
- ↑ H.A. Krebs, W.A. Johnson, Metabolism of ketonic acids in animal tissues, „The Biochemical Journal”, 31 (4), 1937, s. 645–660, DOI: 10.1042/bj0310645, PMID: 16746382, PMCID: PMC1266984 [dostęp 2023-11-28] (ang.).
Bibliografia
- Jeremy Mark Berg, John L Tymoczko, Lubert Stryer, Neil D Clarke, Zofia Szweykowska-Kulińska, Artur Jarmołowski, Halina Augustyniak: Biochemia. Warszawa: Wydawnictwo Naukowe PWN, 2007. ISBN 978-83-01-14379-4.
- B Hames, Nigel M Hooper, Lilla Hryniewiecka, Kazimierz Ziemnicki, Halina Augustyniak: Biochemia. Warszawa: Wydawnictwo Naukowe PWN, 2007. ISBN 978-83-01-13872-1.
- Władysław J. H. Kunicki-Goldfinger: Życie bakterii. Warszawa: Wydawnictwo Naukowe PWN, 1994. ISBN 83-01-11323-5.
- Nicholls, D. G. i Ferguson, S. J., Bioenergetyka 2 Wydawnictwo Naukowe PWN, Warszawa, 1995. ISBN 978-83-01-11661-3
- Lansing M. Prescott: Microbiology. McGraw Hill Higher Education, 2002. ISBN 978-0072485226.
- January Weiner: Życie i ewolucja biosfery. Warszawa: Wydawnictwo Naukowe PWN, 2005. ISBN 83-01-14174-3.
Linki zewnętrzne
- Informacje ogólne
- Metabolism, Cellular Respiration and Photosynthesis. BioChemWeb.org. [dostęp 2009-02-06]. [zarchiwizowane z tego adresu (2014-07-02)]. (ang.). (Metabolizm, oddychanie komórkowe i fotosynteza)
- Biochemistry of Metabolism. Instructional materials for a studio-format course. Rensselaer Polytechnic Institute. [dostęp 2008-11-01]. [zarchiwizowane z tego adresu (22 listopada 2013)]. (ang.). (Biochemia metabolizmu)
- Steyermark A., Njoku R.: Physiology at its best. [dostęp 2008-11-01]. [zarchiwizowane z tego adresu (17 czerwca 2008)]. (ang.). (Zaawansowany kalkulator metabolizmu zwierząt. Interaktywna strona do nauki)
- Hurlbert E.: Microbial metabolism. 1999. [dostęp 2008-11-01]. [zarchiwizowane z tego adresu (24 maja 2013)]. (ang.). (Metabolizm mikroorganizmów)
- Karl J. Miller: Metabolic Pathways of Biochemistry. 1998. [dostęp 2009-02-06]. [zarchiwizowane z tego adresu (2000-08-16)]. (ang.). (Biochemiczne szlaki metaboliczne)
- Chemistry for Biologists. Royal Society of Chemistry, Biochemical Society. [dostęp 2009-02-06]. [zarchiwizowane z tego adresu (2006-04-26)]. (ang.). (Chemia dla biologów)
- Sparknotes: SAT Biology. SparkNotes LLC. [dostęp 2009-02-06]. [zarchiwizowane z tego adresu (2019-03-17)]. (ang.).
- metabolism, [w:] Encyclopædia Britannica [dostęp 2009-02-06] (ang.).
- Metabolizm człowieka
- James Baggott, Sharon E. Dennis: NetBiochem Topics. 1994-95. [dostęp 2009-02-06]. (ang.). (przewodnik nt. szlaków metabolicznych człowieka, poziom szkolny)
- Michael W. King: The Medical Biochemistry Page. [dostęp 2009-02-06]. (ang.). (zasób materiałów nt. metabolizmu człowieka)
- Bazy danych
- The BioCyc Collection of Pathway/Genome Databases (en)
- Flow Chart of Metabolic Pathways (en) (baza danych szlaków metabolicznych)
- The KEGG PATHWAY Database (en) (baza danych szlaków metabolicznych)
- Reactome (baza danych o procesach biologicznych)
- Szlaki metaboliczne
- Donald E. Nicholson: IUBMB-Nicholson Metabolic Pathways Chart. Sigma-Aldrich. [dostęp 2009-02-06]. [zarchiwizowane z tego adresu (2008-09-10)]. (ang.). (Szlaki metaboliczne komórki roślinnej)
- Doutor Pedro Silva: Metabolic pathways. 2002. [dostęp 2009-02-06]. [zarchiwizowane z tego adresu (2013-05-22)]. (ang.). (Schematy głównych szlaków metabolicznych)