| phaeochromocytoma |
Guz chromochłonny (barwiak, łac. phaeochromocytoma) – zazwyczaj łagodny nowotwór wywodzący się najczęściej z komórek chromochłonnych zlokalizowanych w rdzeniu nadnerczy, wydzielających katecholaminy; najczęściej adrenalinę i noradrenalinę. Rzadziej guz chromochłonny powstaje z pozanadnerczowej tkanki chromafinowej, która nie uległa inwolucji po urodzeniu[1]. Jest częstą przyczyną nadciśnienia wtórnego. Guzami o podobnej budowie histologicznej są przyzwojaki.
Terminologia
Guzy chromochłonne wywodzą się z neuroektodermy grzebienia nerwowego. Komórki te występują głównie w rdzeniu nadnerczy, ale również w zwojach i splotach przedkręgowych oraz ciałkach przyzwojowych. Istotą guza chromochłonnego jest wydzielanie katecholamin. Katecholaminy ulegają reakcji z solami kwasu chromowego, stąd komórki je produkujące określa się chromochłonnymi. Natomiast w standardowym barwieniu histopatologicznym nie można odróżnić komórek wydzielających katecholaminy (rdzeń nadnerczy, neurony współczulne) od takich, które nie posiadają tej zdolności (neurony przywspółczulne). Dlatego pod względem histopatologicznym pojęcia pheochromocytoma (guz chromochłonny) i paraganglioma (nerwiak przyzwojowy) można uznać za tożsame. Guzy chromochłonne są więc czynnymi nerwiakami przyzwojowymi.
Guzy chromochłonne rozwijają się najczęściej w rdzeniu nadnerczy, ale mogą również występować w lokalizacji pozanadnerczowej (najczęściej w jamie brzusznej, rzadziej w klatce piersiowej). Nerwiaki przyzwojowe nieczynne hormonalnie (z neuronów przywspółczulnych) zlokalizowane są głównie w obrębie klatki piersiowej, do nich należą również guzy wywodzące się z kłębków pełniących funkcję chemoreceptorów – guzy takowe określa się mianem chemodektoma.
Obecnie proponuje się, żeby stosować określenie guz chromochłonny tylko dla czynnych hormonalnie guzów zlokalizowanych w rdzeniu nadnerczy, natomiast w lokalizacji pozanadnerczowej – nerwiak przyzwojowy współczulny (czynny hormonalnie; dawniej guz chromochłonny pozanadnerczowy) i nerwiak przyzwojowy przywspółczulny (nieczynny hormonalnie).
Epidemiologia
W Stanach Zjednoczonych pheochromocytoma jest obserwowana u 2–8 osób na milion, co daje około 1000 przypadków diagnozowanych każdego roku.
Guz chromochłonny bywa nazywany „nowotworem 10%”[2]:
- W 10% jest zlokalizowany poza nadnerczami (wywodzi się z tkanki chromochłonnej położonej wzdłuż aorty, w okolicy nerek, pęcherza moczowego, wątroby; rzadko nadprzeponowo – wzdłuż aorty piersiowej, w obrębie serca, sporadycznie w obrębie szyi, podstawy czaszki, gruczołu krokowego, powrózka nasiennego). W jednym z badań stwierdzono lokalizację pozanadnerczową w 25% przypadków[3].
- W 10% to nowotwory złośliwe[4] (w przypadku guzów pozanadnerczowych ten odsetek jest wyższy).
- w 10% jest zlokalizowany w obu nadnerczach[3].
- 10% dotyczy dzieci.
- 10% nawraca po resekcji.
- 10% ma charakter mnogi.
- u 10% osób nie występuje nadciśnienie[5] (w jednym z badań stwierdzono, że u 6,1% osób[6]).
Guzy w przebiegu zespołu rodzinnego częściej występują w młodszym wieku i mają mnogi charakter. U dzieci również częściej występuje lokalizacja pozanadnerczowa. Guza chromochłonnego rozpoznaje się wśród 5% wszystkich incydentaloma[5]. Przypadkowe rozpoznania guza chromochłonnego mogą sięgać 30% przy lepszej dostępności do technik obrazowania[6].
Szacuje się, że jest przyczyną około 0,1% przypadków nadciśnienia. Występuje zwykle w 4. i 5. dekadzie życia, ale może również występować zarówno w starszym, jak i młodszym wieku[7].
Etiopatogeneza
Genetyka
Izolowany guz chromochłonny może mieć związek z mutacjami wielu genów, takich jak[8]:
- VHL w locus 3p26-p25
- RET w locus 10q11
- SDHD w locus 11q23
- SDHB w locus 1p36-p35
- KIF1B w locus 1p36.2.
Przypadki związane z uwarunkowaniami genetycznymi występują w zespole mnogiej gruczolakowatości wewnątrzwydzielniczej typu 2A i 2B, nerwiakowłókniakowatości typu 1, zespole von Hippla-Lindaua i zespole guzów chromochłonnych i przyzwojaków (PPS).
Mutacje germinalne identyfikuje się już w około 25% guzów[9].
Patomorfologia
_histopathology.jpg.webp)

Guz chromochłonny zbudowany jest z feochromocytów (komórki chromochłonne) prawidłowo tworzących rdzeń nadnerczy (jest to główne miejsce występowania tych komórek, ale mogą również występować pozanadnerczowo). Są one wieloboczne lub wrzecionowate, brązowo-czarne po barwieniu dwuchromianem potasu, pochodzą z grzebienia nerwowego. Pod wpływem pobudzenia współczulnego wydzielają katecholaminy. Komórki tworzą gniazda (niem. Zellballen) w bogato unaczynionym zrębie.
Makroskopowo guz może być niewielką, otorbioną zmianą w nadnerczu lub nawet kilkukilogramowym, krwotocznym tworem zajmującym duży obszar jamy brzusznej. Zazwyczaj nie przekracza kilku centymetrów, choć opisywano guzy wielkości od kilku milimetrów do 20–30 centymetrów[10]. Przeciętna masa wynosi ok. 100 g[10].
Guzy są zazwyczaj bogato unaczynione, w ich wnętrzu można stwierdzić wylewy krwi, ogniska martwicy, torbiele rzekome i niekiedy odkładanie złogów wapnia.
Niektórzy autorzy z uwagi na budowę histologiczną klasyfikują guzy chromochłonne na 3 typy[10]:
- Guzy nabłonkowe – zbudowane z komórek nabłonkowych, zawierające ziarnistą, kwasochłonną cytoplazmę i hiperchromatyczne jądra; komórki układają się w sznury i skupienia.
- Guzy wielokształtne – zbudowane z komórek nabłonkowych, ale o większym stopniu zróżnicowania (obecne nawet komórki olbrzymie) i większej hiperchromatyczności.
- Guzy wrzecionowate – przeważają komórki wrzecionowate; jest to najrzadsza postać.
Nawet zmiany łagodne mogą naciekać torebkę łącznotkankową i okoliczne naczynia. Na podstawie badania histopatologicznego nie można potwierdzić lub wykluczyć zmiany złośliwej. Złośliwość danej zmiany można stwierdzić tylko na podstawie obecności przerzutów (zazwyczaj do okolicznych węzłów chłonnych, wątroby, płuc, kości, mózgu).
Patofizjologia
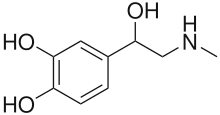

Istotą pheochromocytoma jest nadmierne wydzielanie katecholamin, które powstają w naturalnych, fizjologicznych procesach. Ich prekursorem jest tyrozyna, pobierana przez zakończenia współczulne i następnie metabolizowana pod wpływem hydroksylazy tyrozynowej do 3,4-dihydroksyfenyloalaniny (DOPA). Z Dopy przy udziale dekarboksylazy powstaje dopamina, która ulega dalszym przekształceniom do metoksydopaminy, kwasu 3,4-dihydroksyfenylooctowego (DOPAC), kwasu homowanilinowego (HVA) oraz za pomocą β-hydroksylazy do noradrenaliny (około 25% dopaminy). Pozostała część dopaminy ulega wydaleniu z moczem w postaci wolnej lub związanej. Pod wpływem N-metylotransferazy z noradrenaliny powstaje adrenalina. Guzy zlokalizowane nadprzeponowo nie wydzielają noradrenaliny.
Unieczynnianie noradrenaliny i adrenaliny zachodzi przy udziale monoaminooksydazy (MAO) i katecholo-O-metylotransferazy (COMT). W efekcie ich działania powstają nieaktywne metabolity (metoksykatecholaminy, kwas 3,4-dihydroksymigdałowy, 3-metoksy-hydroksyfenyloglikol, kwas wanilinomigdałowy – istotne w procesie diagnostycznym pheochromocytoma), które są wydalane z moczem. Niewielka część katecholamin zostaje unieczynniona poprzez związanie z kwasem glukuronowym i kwasem siarkowym, a także wychwycona zwrotnie przez zakończenia nerwowe i pozaneuronalne.
Objawy
Głównym objawem guza chromochłonnego jest nadciśnienie tętnicze, które może mieć charakter napadowy lub utrwalony (w rzadkich przypadkach choroba może przebiegać z prawidłowym ciśnieniem). Napady trwają różnie długo (od kilku minut do paru godzin), mogą występować też z różną częstotliwością. Towarzyszą im zwykle silne bóle głowy, zlewne poty, tachykardia, bladość skóry, uczucie niepokoju, zaburzenia czucia, drżenia. U niektórych chorych może występować rozszerzenie źrenic. Po napadzie występuje zwykle znużenie i wyczerpanie.
Napady mogą być wyzwalane wysiłkiem fizycznym, stresem, uciśnięciem brzucha, posiłkiem, zmianą pozycji ciała, długotrwałym przebywaniem w pozycji stojącej, stosunkiem płciowym, defekacją, lekami (histamina, guanetydyna, glukagon, droperidol, metoklopramid, tyramina, trójcykliczne leki przeciwdepresyjne, fenotiazyna, niektóre cytostatyki). Chorzy są zwykle szczupli na skutek zwiększonego metabolizmu.
U niektórych chorych guz chromochłonny może przebiegać z hipotonią ortostatyczną[11]. Może to być spowodowane zaburzeniem mechanizmów współczulnych odpowiedzialnych za regulacje ciśnienia tętniczego przy zmianie pozycji ciała lub zmniejszeniem objętości krwi krążącej.
U pacjentów z guzem umiejscowionym w pęcherzu moczowym może dochodzić do zaburzeń mikcji, krwinkomoczu (napady nadciśnienia mogą również być wywoływane przez oddanie moczu). Niekiedy może dojść do powstania wodonercza jako manifestacji zaburzenia odpływu moczu spowodowanego uciskiem guza[12].
Mogą również pojawiać się dolegliwości bólowe w jamie brzusznej, nudności, wymioty, zaparcia, ostre rozdęcie okrężnicy. Martwiczo zmieniony guz, który uległ pęknięciu, może dawać objawy ostrego brzucha. W przebiegu pheochromocytoma istnieje zwiększone ryzyko kamicy żółciowej[13].
W efekcie oddziaływania katecholamin na mięsień sercowy może dochodzić do zmian o typie kardiomiopatii i w efekcie do powiększenia sylwetki serca, tachykardii, zastoju w krążeniu płucnym. Niekiedy dominują zaburzenia rytmu serca – mogą występować ekstrasystolie nadkomorowe czy komorowe, migotanie przedsionków, napadowy częstoskurcz nadkomorowy, czy nawet migotanie komór. Guz chromochłonny związany jest ze zwiększonym ryzykiem niedokrwienia mięśnia sercowego, udaru mózgowego i uszkodzeń nerek. Istnieje również zagrożenie wystąpieniem nagłego zgonu sercowego.
Niekiedy występują zmiany naczyniowe – mrowienie i drętwienie kończyn czy objawy przypominające zespół Raynauda[14].
Innym objawem który można zaobserwować u chorych z guzem chromochłonnym jest gorączka. Może mieć charakter utrwalony, zwykle nie towarzyszą jej dreszcze. Jest wyrazem zwiększonego metabolizmu i zaburzonej regulacji ciepła (zmniejszona utrata ciepła przez skórę na skutek skurczu naczyń), a także zwiększonego wytwarzania przez guz interleukiny 6[15].
W przebiegu pheochromocytoma może dochodzić do zmian w obrębie układu nerwowego – od parestezji, zaburzeń widzenia, napadów drgawek aż po utratę przytomności. W rzadkich przypadkach może dochodzić do zmian psychicznych.
W podstawowych badaniach laboratoryjnych można niekiedy zaobserwować leukocytozę obojętnochłonną czy policytemię.
Pheochromocytoma ujawniający się w trakcie ciąży może powodować przedwczesne oddzielenie łożyska i poronienie.
Guz chromochłonny może niekiedy wydzielać inne substancje hormonalnie czynne, np. ACTH lub somatostatynę, prowadząc do objawów typowych dla nadprodukcji tych związków.
Diagnostyka
W celu rozpoznania pheochromocytoma oznacza się wolne katecholaminy lub ich metabolity (kwas wanilinomigdałowy VMA i metoksykatecholaminy) w dobowej zbiórce moczu (metoksykatecholaminy można również oznaczać w surowicy). Największą czułością i swoistością cechuje się oznaczenie metoksykatecholamin w moczu lub surowicy. Pozostałe badania mają mniejszą przydatność diagnostyczną.
Następnym etapem jest zlokalizowanie guza za pomocą badań obrazowych. Najczęściej wykonuje się tomografię komputerową lub rezonans magnetyczny. W przypadkach wątpliwych lub w razie niewykrycia guza stosuje się scyntygrafię (z użyciem m-jodobenzyloguanidyny znakowanej jodem) lub PET.
Konieczny jest również dalszy monitoring pod kątem zespołów chorobowych wynikających z mutacji w obrębie genów VHL, RET, SDHB, SDHD.
Próby farmakologiczne
Na skutek upowszechnienia się bezpiecznych prób biochemicznych próby farmakologiczne mają głównie znaczenie historyczne. Stosuje się je zazwyczaj przy niejednoznacznym obrazie klinicznym, jak również w ośrodkach, które nie mają możliwości przeprowadzenia prób biochemicznych. Istotą prób farmakologicznych jest podanie leku i obserwowanie reakcji organizmu w postaci wzrostu lub spadku ciśnienia. Obecnie stosuje się próby z fentolaminą oraz glukagonem. Dawniej używano także histaminy i tyraminy – obecnie mają one już znaczenie historyczne.
- Próba z fentolaminą

Przed wykonaniem próby konieczne jest odstawienie większości leków, zwłaszcza leków hipotensyjnych i uspokajających, które mogą zafałszować wynik testu. W trakcie badania pacjent musi być ściśle monitorowany, istnieje ryzyko gwałtownego spadku ciśnienia.
Próba polega na podaniu dożylnym 2–5 mg fentolaminy w celu zablokowania receptorów α–adrenergicznych. Mierzy się następnie ciśnienie co 30 s przez 10 min. Za dodatnią próbę przyjmuje się spadek ciśnienia skurczowego o co najmniej 35 mm Hg (4,7 kPa) i rozkurczowego o 25 mm Hg (3,3 kPa) w przeciągu 2–3 min od wstrzyknięcia. Spadek ciśnienia utrzymuje się przez około 10 min, a następnie powraca do wartości wyjściowych.
Próba jest stosunkowo bezpieczna, niemniej istnieje ryzyko niepożądanych efektów ubocznych – nadmierny spadek ciśnienia tętniczego ze wstrząsem włącznie, tachykardia, pocenie się, uderzenia gorąca, zaczerwienie skóry, biegunka, bóle brzucha.
- Próba z glukagonem
Jest to próba prowokacyjna, po podaniu glukagonu, na skutek pobudzenia aktywności adenylocyklazy, następuje wzrost stężenia cAMP w komórkach guza i wyrzut katecholamin z gwałtownym wzrostem ciśnienia tętniczego.
Próbę wykonuje się, podając dożylnie 0,5–1 mg glukagonu, a następnie mierzy się ciśnienie co 30 sek przez 5 min, a potem co 60 s przez następne 10 min. Za dodatni wynik uważa się wzrost ciśnienia skurczowego o 60 mm Hg (8,0 kPa) i rozkurczowego o 30 mm Hg (4,0 kPa). Normalizacja ciśnienia następuje zwykle po około 5–15 min od podania glukagonu.
Tuż przed badaniem pobiera się krew i mocz na oznaczenie stężenia katecholamin, a następnie tuż po wstrzyknięciu glukagonu – krew, a 3 h po badaniu – mocz.
Diagnostyka obrazowa

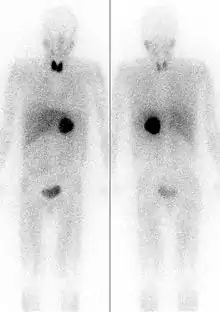
Ustalenie lokalizacji guza jest konieczne celem dobrania odpowiedniej techniki operacyjnej (operacja jest metodą leczenia z wyboru guza chromochłonnego).
Jako metodę przesiewowego badania w kierunku obecności pheochromocytoma poleca się USG jamy brzusznej. Jest to metoda szeroko dostępna i tania. Jednak z uwagi na stosunkowo małą czułość w wykrywaniu małych guzów oraz zlokalizowanych pozanadnerczowo nie może być metodą rozstrzygającą. Całkowitą czułość USG szacuje się na około 40–95%[16].
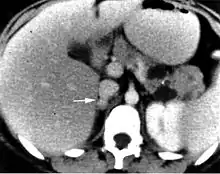
Tomografia komputerowa (TK) jest badaniem ogólnie dostępnym, charakteryzującym się dużą czułością (95–98%[16]). Guzy chromochłonne w obrazie TK charakteryzują się zwykle jednorodną strukturą i owalnym kształtem. Niekiedy można uwidocznić zmiany torbielowate, ogniska martwicy, wylewy, zwapnienia w świetle guza. Przy złośliwych postaciach pheochromocytoma istotna jest również możliwość zlokalizowania potencjalnych przerzutów.
Przy trudnościach diagnostycznych, zwłaszcza w przypadku guzów zlokalizowanych pozanadnerczowo, stosuje się rezonans magnetyczny. Uważa się, że właśnie w przypadku takiej lokalizacji rezonans wykazuje większą czułość. Przydatny jest również w różnicowaniu innych zmian w nadnerczach. Minusem metody jest mniejsza dostępność i swoistość oraz wyższy koszt.
Badaniem charakteryzującym się dużą czułością (sięgającą 100%, przy swoistości 70–80%[16]) jest scyntygrafia z użyciem m-jodobenzyloguanidyny znakowanej radiojodem (mIBG). Komórki o utkaniu chromochłonnym wychwytują MIBG, umożliwiając zlokalizowanie nawet bardzo małych zmian. Badanie jest szczególnie pomocne w wykrywaniu zmian pozanadnerczowych oraz przerzutów.
W rzadkich przypadkach, przy trudnościach diagnostycznych, stosuje się cewnikowanie żyły głównej dolnej, pobierając krew na oznaczenie katecholamin z różnych poziomów. Jest to badanie inwazyjne, obarczone ryzykiem powikłań, rzadko wykonywane.
W specyficznych przypadkach guza umiejscowionego w obrębie klatki piersiowej pomocne może być badanie RTG oraz badanie echokardiograficzne.
Różnicowanie
W diagnostyce różnicowej guza chromochłonnego należy wziąć pod uwagę:
- Nadciśnienie pierwotne – może sprawiać najwięcej trudności diagnostycznych. Nadciśnienie pierwotne może imitować objawy występujące u chorych z guzem chromochłonnym; również sam pheochromocytoma może przebiegać skąpoobjawowo lub w sposób niecharakterystyczny. Dodatkowo u pacjentów z nadciśnieniem pierwotnym i wzmożoną aktywnością układu współczulnego może dochodzić do nieznacznie wzmożonego wydzielania katecholamin i ich metabolitów w moczu. U pacjentów z nadciśnieniem pierwotnym w trakcie napadów ciśnienie tętnicze zwykle nie osiąga tak wysokich wartości jak w przypadku napadów związanych z pheochromocytoma. Skóra jest zwykle zaczerwieniona, a pocenie ograniczone do dłoni, stóp i okolic pachowych. Ostateczne rozpoznanie stawia się na podstawie dodatnich prób biochemicznych.
- Cukrzycę ze współistniejącym nadciśnieniem. W przebiegu pheochromocytoma może dochodzić do różnych zaburzeń gospodarki węglowodanowej, z pełnoobjawową cukrzycą włącznie. U chorych z nadciśnieniem wtórnym do cukrzycy (w przebiegu cukrzycowej chorobie nerek) obserwuje się charakterystyczne zmiany na dnie oka, w badaniu moczu czy obrzęki obwodowe. W przebiegu pheochromocytoma rzadko dochodzi do rozwoju kwasicy. Usunięcie guza zwykle prowadzi do normalizacji glikemii. Ostateczne rozpoznanie stawia się na podstawie dodatnich prób biochemicznych.
- Nadczynność tarczycy – objawy mogą imitować guza chromochłonnego (wzrost ciśnienia, nadmierne pocenie się, tachykardia, niepokój, chudnięcie). Za nadczynnością tarczycy przemawiają: obecność wola, charakterystyczne objawy oczne, zwiększenie poziomu hormonów tarczycowych, prawidłowe stężenie katecholamin i ich metabolitów w moczu. W przebiegu nadczynności tarczycy zwyżki ciśnienie tętniczego dotyczą zazwyczaj ciśnienia skurczowego, nie mają charakteru napadowego i rzadko osiągają wysokie wartości.
- Częstoskurcz napadowy. W trakcie napadu częstoskurczu związanego z guzem chromochłonnym występuje wzrost ciśnienia (w zależnym od innych przyczyn ciśnienie jest zwykle prawidłowe lub obniżone), a także inne objawy związane z nadmiernym wyrzutem katecholamin. Ostateczne rozpoznanie potwierdza oznaczenie katecholamin w moczu zebranym bezpośrednio po napadzie.
- Chorobę niedokrwienną serca. W trakcie napadu silnych bólów stenokardialnych może dochodzić do wzrostu ciśnienia z tachykardią, zblednięciem powłok i niepokojem. O rozpoznaniu przesądza obraz kliniczny oraz oznaczenie poziomu katecholamin w moczu (choć u pacjentów z silnymi bólami, czy w pierwszej dobie zawału może dochodzić do wzrostu stężenia katecholamin[17] to wartości te są niższe niż u pacjentów z guzem chromochłonnym).
- Guzy mózgu, zwłaszcza zlokalizowane w tylnej jamie czaszkowej – mogą przebiegać z wysokim ciśnieniem i innymi objawami pobudzenia układu współczulnego (również ze zwiększeniem stężenia katecholamin w moczu). Charakterystyczne jedynie dla guzów mózgu są specyficzne objawy neurologiczne oraz nieprawidłowości w badaniach obrazowych głowy (choć w rzadkich przypadkach guz chromochłonny może być umiejscowiony w centralnym systemie nerwowym). Częściej występuje też bradykardia.
- Bóle głowy o charakterze migrenowym, klasterowym czy naczynioruchowym – mogą przebiegać ze wzrostem ciśnienia i objawami pobudzenia układu współczulnego.
- Rodzinną dysautonomię i zespół Guillaina-Barrégo – mogą przebiegać z wysokim ciśnieniem tętniczym, nadmiernym poceniem się, czy niepokojem. Opisywano przypadki zwiększonego stężenia katecholamin i ich metabolitów w osoczu i moczu u pacjentów z zespołem Guillaina-Barrégo[18]. Różnicowanie z guzem chromochłonnym polega na stwierdzeniu charakterystycznej polineuropatii z osłabieniem siły mięśniowej.
- Przekwitanie u kobiet – stosunkowo często występują zwyżki ciśnienia, objawy naczynioruchowe, nadmierna potliwość. Dość charakterystycznym objawem dla przekwitania różnicującym go z guzem chromochłonnym jest duża skłonność do zaczerwienienia skóry. Ostateczne rozpoznanie stawia się na podstawie dodatnich prób biochemicznych.
- Ostrą przerywana porfirię w obrazie której może występować nadciśnienie i tachykardia, a także zwiększone wydalanie katecholamin z moczem[19]. Rozpoznanie stawia się na podstawie objawów klinicznych (bóle brzucha, objawy neurologiczne, ciemne zabarwienie moczu), a także stwierdzenia zwiększonego wydalania porfobilinogenu i kwasu aminolewulinowego.
- Pseudopheochromocytoma – jest to stan w którym występują charakterystyczne objawy guza chromochłonnego, a nie stwierdza się dodatnich prób biochemicznych i nie udało się zobrazować guza. Sugeruje się, że może on wynikać z zaburzenia wiązania katecholamin[20].
- Zespół hiperkinetyczny spowodowany nadmierną aktywnością receptorów β-adrenergicznych.
- Nadciśnienie naczyniowonerkowe.
- Przyzwojaki.
- Zaburzenia lękowe.
Leczenie
Leczenie farmakologiczne
| Lek | Dawka [mg/d] |
|---|---|
| α-blokery | |
| Fenoksybenzamina | 30–100 |
| Prazosyna | 3–15 |
| Doksazosyna | 1–16 |
| Terazosyna | 1–20 |
| β-blokery | |
| Propranolol | 60–120 |
| Atenolol | 25–100 |
| Metoprolol | 50–100 |
| α- i β-blokery | |
| Labetalol | 300–1200 |
Przed planowanym zabiegiem operacyjnym, który jest leczeniem z wyboru u pacjentów z guzem chromochłonnym, konieczne jest odpowiednie przygotowanie farmakologiczne. Ma ono na celu przywrócenie prawidłowego ciśnienia, zapobieżenie jego gwałtownym skokom podczas operacji oraz normalizację akcji serca. Zazwyczaj okres przygotowawczy wynosi około 2 tygodni[22], przy silniej wyrażonych objawach klinicznych, czy współistnieniu dodatkowych powikłań ze strony układu sercowo-naczyniowego może ulec on wydłużeniu.
Lekami preferowanymi są α-blokery. Stosuje je się do momentu uzyskania prawidłowego ciśnienia krwi, bez wahań w ciągu dnia. Zazwyczaj podaje się doustnie fenoksybenzaminę, zaczynając od mniejszej dawki i stopniowo ją zwiększając do uzyskania pełnego efektu terapeutycznego. Można również stosować inne α-blokery, także nowszej generacji (istnieje jednak mniej badań klinicznych potwierdzających ich skuteczność). Przeważa opinia, że leki alfa-adrenolityczne stwarzają najlepsze warunki do przeprowadzenia operacji, choć część autorów uważa, że mogą utrudnić manualne wykrycie guza podczas zabiegu operacyjnego, oraz prowadzić do nadmiernych spadków ciśnienia w okresie pooperacyjnym.
Jako leki uzupełniające stosuje się β-blokery, zwłaszcza u osób ze współistniejącą tachykardią czy zaburzeniami rytmu serca (napadowe częstoskurcze, dodatkowe pobudzenia nadkomorowe). Są one niewystarczające w monoterapii w okresie przygotowawczym, gdyż mogą sprzyjać generowaniu nadciśnienia z uwagi na zwiększone powinowactwo katecholamin do niezablokowanych receptorów α[23]. Powinny być stosowane wyłącznie w połączeniu z α-blokerami[24].
Labetalol, blokujący zarówno receptory β-adrenergiczne, jak i α-adrenergiczne, nie powinien być traktowany jako lek pierwszego rzutu, gdyż stosunek blokady receptorów β do blokady receptorów α (przyjmowany doustnie) wynosi 7:1[25] – po jego podaniu może dojść do paradoksalnego wzrostu ciśnienia tętniczego w mechanizmie opisanym powyżej[26].
Niekiedy stosuje się również blokery kanałów wapniowych, w niektórych ośrodkach są one nawet preferowanym lekiem u pacjentów z guzem chromochłonnym i prawidłowym ciśnieniem tętniczym[27]. Zazwyczaj jednak stosuje się je w terapii łączonej z α-blokerami – przy niedostatecznej kontroli ciśnienia lub w monoterapii – przy występowaniu efektów ubocznych skutkujących koniecznością odstawienia α-blokerów.
Bardzo ważnym elementem przygotowania do zabiegu jest uzupełnienie niedoborów objętości krążącej krwi, które to występują dość często u pacjentów z guzem chromochłonnym. Niekiedy istnieje konieczność przetoczenia krwi.
Leczenie chirurgiczne

Jedyną metodą skutecznego leczenia guza chromochłonnego jest jego chirurgiczne usunięcie. Jest to metoda leczenia z wyboru w tym schorzeniu. Jedynie w nielicznych przypadkach odstępuje się od zabiegu (bezwzględne przeciwwskazania do operacji, brak zgody pacjenta, złośliwy guz z uogólnionymi przerzutami).
Zabieg polega na usunięciu guza. Najczęściej jest on zlokalizowany w nadnerczach – stosuje się wtedy adrenalektomię (usunięcie nadnercza). Ostatnio coraz większe uznanie zyskuje metoda laparoskopowa[28]. Możliwa jest również operacja drogą laparotomii[29]. W wybranych przypadkach możliwe jest zastosowanie adrenalektomii oszczędzającej – pozwala na zachowanie prawidłowej funkcji kory nadnerczy.
Zabieg operacyjny u pacjentów z guzem chromochłonnym wymaga modyfikacji środka znieczulającego. W premedykacji preferowane są pochodne benzodiazepin (nie działają na układ współczulny). W znieczuleniu ogólnym stosuje się podtlenek azotu, enfluran, izofluran, fentanyl i izofentanyl. Nie poleca się halotanu, który może nasilać zaburzenia rytmu serca.
W trakcie zabiegu napadowe zwyżki ciśnienia tętniczego opanowuje się dożylnym podaniem 2–3 mg fentolaminy lub nitroprusydkiem sodu (we wlewie dożylnym 0,25–10 μg/kg m.c./min) czy nitrogliceryną (5–100 μg/min). W zaburzeniach rytmu serca stosuje się dożylnie propranolol (5–10 mg), lidokainę (50–100 mg) czy esmolol (200–500 μg/kg m.c./min). Nadmierny spadek ciśnienia jest wskazaniem do wypełnienia łożyska naczyniowego (podanie krwi lub innych płynów) lub w rzadkich przypadkach zastosowania katecholamin (np. noradrenaliny). Bezpośrednio po usunięciu guza może dojść do hipoglikemii (zniesienie hamującego wpływu katecholamin na sekrecję insuliny).
Po zabiegu operacyjnym konieczne jest monitorowanie pacjenta pod kątem ciśnienia tętniczego, elektrokardiogramu i tętna. Utrzymywanie się wysokiego ciśnienia po operacji może świadczyć o obecności drugiego guza chromochłonnego lub czynnych hormonalnie przerzutów, współistnieniu nadciśnienia pierwotnego (lub występowania utrwalonych zmian naczyniowych) czy, w rzadkich przypadkach, powikłania pooperacyjnego w postaci uszkodzenia tętnicy nerkowej. W każdym przypadku należy oznaczyć stężenie katecholamin lub ich metabolitów w moczu. Niekiedy konieczna jest substytucja steroidów (przy obustronnym usunięciu nadnerczy).
Niezbędna jest długoterminowa kontrola pacjentów po usunięciu guza chromochłonnego polegająca na monitorowaniu ciśnienia tętniczego oraz okresowym oznaczaniu katecholamin i ich metabolitów w moczu.
Leczenie złośliwego guza chromochłonnego
Nieleczony złośliwy guz chromochłonny w około 50% przypadków prowadzi do zgonu w przeciągu 5 lat[30]. Nie ma skutecznej metody leczącej złośliwą postać guza chromochłonnego. W leczeniu chirurgicznym należy dążyć do usunięcia jak największej ilości aktywnej hormonalnie tkanki, zarówno w obrębie guza pierwotnego, jak i przerzutów. Można w ten sposób osiągnąć spowolnienie choroby i zmniejszyć nasilenie objawów klinicznych poprzez zmniejszenie objętości krążących katecholamin[31].
Metodami alternatywnymi do zabiegu operacyjnego są: radioterapia, krioablacja, TAE (transcatheter arterial embolization), ablacja przezskórna czy chemioterapia[32][33]. W skojarzonej chemioterapii wykorzystuje się cyklofosfamid, winkrystynę i dakarbazynę. Terapia taka może prowadzić u około 50% chorych do przejściowej remisji[34]. Stosuje się również m-jodobenzyloguanidynę w dawkach przekraczających te stosowane w celach diagnostycznych[35][36][37].
U pacjentów z wysokim poziomem krążących katecholamin skuteczna może się okazać terapia przy użyciu α-metylotyrozyny[38][39]. Jest to związek podobny do 1-tyrozyny, który hamuje przekształcanie tyrozyny do dihydroksyfenyloalaniny (DOPA), a co za tym idzie powstawanie katecholamin. Stosowanie tego związku jest jednak ograniczone z powodu stosunkowo częstych efektów ubocznych.
Wśród potencjalnych leków mogących mieć zastosowanie w terapii złośliwego guza chromochłonnego wymienia się analogi somatostatyny[40]. Część guzów neuroendokrynnych posiada na powierzchni swoich komórek różne typy receptorów somatostatynowych, wobec czego leki takie mogą okazać się skuteczną metodą terapii. Pojedyncze badania donosiły o redukcji produkcji katecholamin przez komórki guza po zastosowaniu oktreotydu[41], natomiast pozostałe sugerowały jego małą przydatność[42].
Zwiększona ekspresja białka szoku cieplnego HSP90 w złośliwych guzach chromochłonnych[43] sugeruje zastosowanie jego inhibitorów w terapii[44].
Rokowanie
5-letni okres przeżycia w przypadku guza łagodnego wynosi 96%, natomiast przy guzie złośliwym 44%. Ryzyko nawrotu guza łagodnego sięga około 10%. U pacjentów z guzem chromochłonnym istnieje zwiększona śmiertelność związana z obecnością dodatkowego guza (innego niż pheochromocytoma)[45][46].
Śmiertelność okołooperacyjna nie przekracza 3%. U około 60% pacjentów po zabiegu operacyjnym dochodzi do normalizacji ciśnienia tętniczego. Częściej normalizacja dotyczy pacjentów z napadowym nadciśnieniem (około 80%) niż z utrwalonym (około 40%)[47].
Historia
W 1886 Felix Fränkel jako pierwszy opisał guza chromochłonnego u 18-letniej chorej[48][49]. Paul Manasse w 1893 dokonał opisu anatomopatologicznego guza[50]. W 1904 G. Marchetii opisał obustronny guz nadnerczy[51]. W 1908 roku Henri Alezais i Felix Peyron wprowadzili termin paraganglioma. Ludwig Pick w 1912 zauważył powinowactwo komórek guza do soli chromu – do nomenklatury medycznej został wprowadzony termin pheochromocytoma[52]. Możliwość współistnienia guza chromochłonnego z neurofibromatosis odkrył w 1910 S. Suzuki[53]. W 1922 M. L’Abbé ze współpracownikami dokonał pełnego klinicznego opisu pheochromocytoma[54].
W 1926 César Roux w Szwajcarii[55] i w 1927 Charles Horace Mayo w Stanach Zjednoczonych wykonali pierwszy zabieg adrenalektomii u pacjenta z guzem chromochłonnym[56]. Pierwszy zabieg usunięcia guza chromochłonnego rozpoznanego przedoperacyjnie przeprowadził w 1929 Maurice C. Pincoffs[57]. W tym samym roku Coleman Berley Rabin wykazał obecność czynnika presyjnego w guzie[58], który w 1935 został zidentyfikowany jako adrenalina[59].
W 1943 A. Hyman i W.H. Mencher opisali przypadek rodzinnego występowania guza chromochłonnego[60]. Rok 1945 to wprowadzenie testu z histaminą[61]. A. Engel i Ulf Svante von Euler w 1950 odkryli metodę oznaczania katecholamin w moczu[62]. W 1956 H. Weil-Malherbe odkrył w guzie chromochłonnym obecność dihydroksyfenyloalaniny (DOPA)[63]. W 1957 Marvin D. Armstrong ze współpracownikami wykazali zwiększoną zawartość kwasu wanilinomigdałowego w moczu osób chorych na pheochromocytoma, stwierdzając jednocześnie, że jest on głównym metabolitem adrenaliny i noradrenaliny[64].
Związek guza chromochłonnego z rakiem rdzeniastym tarczycy zauważył w 1961 John H. Sipple[65]. W 1965 M. Lawrence wprowadził do diagnostyki test z glukagonem[66]. Przydatność tomografii komputerowej w diagnostyce guza wykazał Stewart ze współpracownikami w 1978[67]. W 1981 Sisson ze współpracownikami wprowadzili m-jodobenzyloguanidynę do diagnostyki[68].
W 1993 zostały opublikowane wyniki badań nad częstością występowania guza chromochłonnego w zespole MEN 2[69]. Stały się podstawą do stwierdzenia konieczności przeprowadzania badań u pacjentów z guzem chromochłonnym pod kątem współwystępowania raka rdzeniastego tarczycy i nadczynności przytarczyc.
Klasyfikacja ICD10
| kod ICD10 | nazwa choroby |
|---|---|
| ICD-10: C74.1 | Nowotwór złośliwy rdzenia nadnercza |
| ICD-10: D35.0 | Nowotwór niezłośliwy nadnercza |
Przypisy
- ↑ Walter F. Boron, Emile L. Boulpaep: Medical physiology: a cellular and molecular approach. Philadelphia, Pa.: W.B. Saunders, 2003. ISBN 0-7216-3256-4.
- ↑ Vinay Kumar, Ramzi S. Cotran, Stanley L. Robbins: Robbins basic pathology. Philadelphia: Saunders, 2003. ISBN 0-7216-9274-5.
- 1 2 Madani R., Al-Hashmi M., Bliss R., Lennard T.W. Ectopic pheochromocytoma: does the rule of tens apply?. „World journal of surgery”. 4 (31), s. 849–854, 2007. DOI: 10.1007/s00268-006-0608-1. PMID: 17372668.
- ↑ Adjallé R., Plouin P.F., Pacak K., Lehnert H. Treatment of Malignant Pheochromocytoma. „Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme”, 2009. DOI: 10.1055/s-0029-1231025. PMID: 19672813.
- 1 2 Alan J. Wein, Meredith F. Campbell, Patrick C. Walsh, Louis R. Kavoussi, Andrew C. Novick, Alan W. Partin, Craig Peters: Campbell-Walsh urology. Philadelphia: W.B. Saunders, 2007. ISBN 978-0-7216-0798-6.
- 1 2 Kopetschke R., Slisko M., Kilisli A., Tuschy U., Wallaschofski H., Fassnacht M., Ventz M., Beuschlein F., Reincke M., Reisch N., Quinkler M. Frequent incidental discovery of phaeochromocytoma: data from a German cohort of 201 phaeochromocytoma. „European journal of endocrinology / European Federation of Endocrine Societies”. 2 (161), s. 355–361, 2009. DOI: 10.1530/EJE-09-0384. PMID: 19497985.
- ↑ Andrzej Szczeklik: Choroby wewnętrzne. Kraków: Medycyna Praktyczna, 2005, s. 1126. ISBN 83-7430-031-0.
- ↑ Maher E.R, Eng C. The pressure rises: update on the genetics of phaeochromocytoma. „Human molecular genetics”. 20 (11), s. 2347–2354, 2002. PMID: 12351569.
- ↑ Gerd Herold: Medycyna Wewnętrzna. Warszawa: PZWL, 2008, s. 387. ISBN 978-83-200-3942-9.
- 1 2 3 Anatomia patologiczna. W: Włodzimierz Januszewicz, Bożenna Wocial, Marek Sznajderman, Andrzej Januszewicz: Guz chromochłonny. Wyd. II. Warszawa: Wydawnictwo Lekarskie PZWL, 2000, s. 31–35. ISBN 83-200-2344-0.
- ↑ D.E. Gennes L., Bricaire H., Moreau L., Courjaret J., Blanc G., Pasquier P. [Pheochromocytoma with true orthostatic hypotension. Report of a case and review of the literature]. „Bulletins et mémoires de la Société médicale des hôpitaux de Paris”, s. 1035–1048, 1963. PMID: 14098864.
- ↑ Martín Osés E., Páez Borda A., Luján Galán M., Ruiz de la Roja J.C., Sánchez Sánchez E., Berenguer Sánchez A. [Bladder pheochromocytoma: a rare cause of ureterohydronephrosis]. „Archivos españoles de urología”. 8 (50), s. 915–917, 1997. PMID: 9463292.
- ↑ Gluvić Z., Rasić-Milutinović Z. [Pheochromocytoma associated with cholelithiasis]. „Medicinski pregled”. 7–8 (54). s. 383–386. PMID: 11905191.
- ↑ Tsai J.J., Tsai W.J., Yen J.H., Chen J.R., Lin S.F., Liu H.W. Malignant pheochromocytoma associated with Jaccoud’s-type arthropathy, Raynaud’s phenomenon, positive antinuclear antibody and rheumatoid factor. „Gaoxiong yi xue ke xue za zhi = The Kaohsiung journal of medical sciences”. 9 (10), s. 518–521, 1994. PMID: 7983696.
- ↑ Suzuki K., Miyashita A., Inoue Y., Iki S., Enomoto H., Takahashi Y., Takemura T. Interleukin-6-producing pheochromocytoma. „Acta haematologica”. 4 (85), s. 217–219, 1991. PMID: 1853686.
- 1 2 3 Ustalenie lokalizacji guza. W: Włodzimierz Januszewicz, Bożenna Wocial, Marek Sznajderman, Andrzej Januszewicz: Guz chromochłonny. Wyd. II. Warszawa: Wydawnictwo Lekarskie PZWL, 2000, s. 122–127. ISBN 83-200-2344-0.
- ↑ Little R.A., Frayn K.N., Randall P.E., Stoner H.B., Morton C., Yates D.W., Laing G.S. Plasma catecholamines in the acute phase of the response to myocardial infarction. „Archives of emergency medicine”. 1 (3), s. 20–27, 1986. PMID: 3524599.
- ↑ Ahmad J., Kham A.S., Siddiqui M.A. Estimation of plasma and urinary catecholamines in Guillain-Barré syndrome. „Japanese journal of medicine”. 1 (24), s. 24–29, 1985. PMID: 3999461.
- ↑ Beal M.F., Atuk N.O., Westfall T.C., Turner S.M. Catecholamine uptake, accumulation, and release in acute porphyria. „The Journal of clinical investigation”. 5 (60), s. 1141–1148, 1977. DOI: 10.1172/JCI108866. PMID: 908757.
- ↑ Blum I., Weinstein R., Sztern M., Lahav M. Adrenergic receptor hyperactivity--a cause for pseudopheochromocytoma?. „Medical hypotheses”. 1 (22), s. 89–96, 1987. PMID: 3031435.
- ↑ Przygotowanie do zabiegu operacyjnego. W: Włodzimierz Januszewicz, Bożenna Wocial, Marek Sznajderman, Andrzej Januszewicz: Guz chromochłonny. Wyd. II. Warszawa: Wydawnictwo Lekarskie PZWL, 2000. ISBN 83-200-2344-0.
- ↑ van der Horst-Schrivers A.N., Kerstens M.N., Wolffenbuttel B.H. Preoperative pharmacological management of phaeochromocytoma. „The Netherlands journal of medicine”. 8 (64), s. 290–295, 2006. PMID: 16990692.
- ↑ Sloand E.M., Thompson B.T. Propranolol-induced pulmonary edema and shock in a patient with pheochromocytoma. „Archives of internal medicine”. 1 (144), s. 173–174, 1984. PMID: 6691755.
- ↑ Sibal L., Jovanovic A., Agarwal S.C., Peaston R.T., James R.A., Lennard T.W., Bliss R., Batchelor A., Perros P. Phaeochromocytomas presenting as acute crises after beta blockade therapy. „Clinical endocrinology”. 2 (65), s. 186–190, 2006. DOI: 10.1111/j.1365-2265.2006.02571.x. PMID: 16886958.
- ↑ Kanto J.H. Current status of labetalol, the first alpha- and beta-blocking agent. „International journal of clinical pharmacology, therapy, and toxicology”. 11 (23), s. 617–628, 1985. PMID: 2867049.
- ↑ Feek C.M., Earnshaw P.M. Hypertensive response to labetalol in phaeochromocytoma. „British medical journal”. 6236 (281), s. 387, 1980. PMID: 7427286.
- ↑ Ulchaker J.C., Goldfarb D.A., Bravo E.L., Novick A.C. Successful outcomes in pheochromocytoma surgery in the modern era. „The Journal of urology”. 3 (161), s. 764–767, 1999. PMID: 10022680.
- ↑ Tiberio G.A., Baiocchi G.L., Arru L., Agabiti Rosei C., De Ponti S., Matheis A., Rizzoni D., Giulini S.M. Prospective randomized comparison of laparoscopic vesrsus open adrenalectomy for sporadic pheochromocytoma. „Surgical endoscopy”. 6 (22), s. 1435–1439, 2008. DOI: 10.1007/s00464-008-9904-1. PMID: 18398641.
- ↑ Jaroszewski D.E., Tessier D.J., Schlinkert R.T., Grant C.S., Thompson G.B., van Heerden J.A., Farley D.R., Smith S.L., Hinder R.A. Laparoscopic adrenalectomy for pheochromocytoma. „Mayo Clinic proceedings. Mayo Clinic”. 12 (78), s. 1501–1504, 2003. PMID: 14661679.
- ↑ John H., Ziegler W.H., Hauri D., Jaeger P. Pheochromocytomas: can malignant potential be predicted?. „Urology”. 4 (53), s. 679–683, 1999. PMID: 10197840.
- ↑ Mishra A.K., Agarwal G., Kapoor A., Agarwal A., Bhatia E., Mishra S.K. Catecholamine cardiomyopathy in bilateral malignant pheochromocytoma: successful reversal after surgery. „International journal of cardiology”. 1 (76), s. 89–90, 2000. PMID: 11121600.
- ↑ Takahashi K., Ashizawa N., Minami T., Suzuki S., Sakamoto I., Hayashi K., Tomiyasu S., Sumikawa K., Kitamura K., Eto T., Yano K. Malignant pheochromocytoma with multiple hepatic metastases treated by chemotherapy and transcatheter arterial embolization. „Internal medicine (Tokyo, Japan)”. 4 (38), s. 349–354, 1999. PMID: 10361908.
- ↑ Pacak K., Fojo T., Goldstein D.S., Eisenhofer G., Walther M.M., Linehan W.M., Bachenheimer L., Abraham J., Wood B.J. Radiofrequency ablation: a novel approach for treatment of metastatic pheochromocytoma. „Journal of the National Cancer Institute”. 8 (93), s. 648–649, 2001. PMID: 11309443.
- ↑ Averbuch S.D., Steakley C.S., Young R.C., Gelmann E.P., Goldstein D.S., Stull R., Keiser H.R. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. „Annals of internal medicine”. 4 (109), s. 267–273, 1988. PMID: 3395037.
- ↑ Safford S.D., Coleman R.E., Gockerman J.P., Moore J., Feldman J.M., Leight G.S., Tyler D.S., Olson J.A. Iodine -131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytoma and paraganglioma. „Surgery”. 6 (134), s. 956–962; discussion 962–3, 2003. DOI: 10.1016/S0039. PMID: 14668728.
- ↑ Rose B., Matthay K.K., Price D., Huberty J., Klencke B., Norton J.A., Fitzgerald P.A. High-dose 131I-metaiodobenzylguanidine therapy for 12 patients with malignant pheochromocytoma. „Cancer”. 2 (98), s. 239–248, 2003. DOI: 10.1002/cncr.11518. PMID: 12872341.
- ↑ Loh K.C., Fitzgerald P.A., Matthay K.K., Yeo P.P., Price D.C. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. „Journal of endocrinological investigation”. 11 (20), s. 648–658, 1997. PMID: 9492103.
- ↑ Decoulx M., Wemeau J.L., Racadot-Leroy N., Grimbert I., Proye C., Plane C. [Alpha-methyl-paratyrosine in the treatment of malignant pheochromocytoma]. „La Revue de médecine interne / fondée ... par la Société nationale francaise de médecine interne”. 4 (8). s. 383–388. PMID: 3423477.
- ↑ Lehnert H., Mundschenk J., Hahn K. Malignant pheochromocytoma. „Frontiers of hormone research”, s. 155–162, 2004. PMID: 14674310.
- ↑ Wiseman G.A., Kvols L.K. Therapy of neuroendocrine tumors with radiolabeled MIBG and somatostatin analogues. „Seminars in nuclear medicine”. 3 (25), s. 272–278, 1995. PMID: 7570046.
- ↑ Invitti C., De Martin I., Bolla G.B., Pecori Giraldi F., Maestri E., Leonetti G., Cavagnini F. Effect of octreotide on catecholamine plasma levels in patients with chromaffin cell tumors. „Hormone research”. 4 (40), s. 156–160, 1993. PMID: 8300064.
- ↑ Lamarre-Cliche M., Gimenez-Roqueplo A.P., Billaud E., Baudin E., Luton J.P., Plouin P.F. Effects of slow-release octreotide on urinary metanephrine excretion and plasma chromogranin A and catecholamine levels in patients with malignant or recurrent phaeochromocytoma. „Clinical endocrinology”. 5 (57), s. 629–634, 2002. PMID: 12390337.
- ↑ Boltze C., Mundschenk J., Unger N., Schneider-Stock R., Peters B., Mawrin C., Hoang-Vu C., Roessner A., Lehnert H. Expression profile of the telomeric complex discriminates between benign and malignant pheochromocytoma. „The Journal of clinical endocrinology and metabolism”. 9 (88), s. 4280–4286, 2003. PMID: 12970299.
- ↑ Maloney A., Workman P. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. „Expert opinion on biological therapy”. 1 (2), s. 3–24, 2002. DOI: 10.1517/14712598.2.1.3. PMID: 11772336.
- ↑ Khorram-Manesh A., Jansson S., Wängberg B., Nilsson O., Tisell L.E., Ahlman H. Mortality associated with pheochromocytoma: increased risk for additional tumors. „Annals of the New York Academy of Sciences”, s. 444–448, 2006. DOI: 10.1196/annals.1353.048. PMID: 17102113.
- ↑ Khorram-Manesh A., Ahlman H., Nilsson O., Odén A., Jansson S. Mortality associated with pheochromocytoma in a large Swedish cohort. „European journal of surgical oncology: the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology”. 5 (30), s. 556–559, 2004. DOI: 10.1016/j.ejso.2004.03.006. PMID: 15135486.
- ↑ Pruszczyk P., Januszewicz W., Feltynowski T., Chodakowska J., Wocial B., Pachocki R., Nielubowicz J., Szostek M. Long term follow-up after surgical removal of pheochromocytoma--observations in 61 patients. „Clinical and experimental hypertension. Part A, Theory and practice”. 6–7 (13), s. 1179–1194, 1991. PMID: 1760886.
- ↑ Fränkel F. Ein Fall von doppelseitigem, völlig latent verlaufenen Nebennierentumor und gleichzeitiger Nephritis mit Veränderungen am Circulationsapparat und Retinitis. „Arch Pathol Anat Physiol Klin Med”. 103, s. 244–263, 1886.
- ↑ Fränkel F. Classics in oncology. A case of bilateral completely latent adrenal tumor and concurrent nephritis with changes in the circulatory system and retinitis: Felix Fränkel, 1886. „CA: a cancer journal for clinicians”. 2 (34). s. 93–106. PMID: 6423225.
- ↑ Manasse P. Über die hyperplastischen Tumoren der Nebennieren. „Virchow’s Arch Pathol Anat Klin Med”. 133, s. 391–404, 1893.
- ↑ Marchetti G. Beitrag zur Kenntnis der pathologischen Anatomie der Nebennieren. „Virchow. Arch. Pathol. Anat.”. 177, s. 227, 1904.
- ↑ Pick L. Das Ganglioma Embryonale Sympathicum (Sympathoma Embryonale), eine typische bösartige Geschwuestform des sympathischen Nervensystems. „Berl Klin Wochenschr.”. 49, s. 16–22, 1912.
- ↑ Suzuki S. Über Zwei Tumoren aus Nebennierenmarkgewebe. „Berlin Klin Wochenschr.”. 47, s. 1623–1625, 1910.
- ↑ L’Abbé, M., Tinel, J., Doumer, A. Crises solaires et hypertension paroxystique en rapport avec une tumeur surrénale. „Bull. Mém. Soc. Méd. Hôp.”. 46, s. 982, 1922.
- ↑ Welbourn R.B. Early surgical history of phaeochromocytoma. „The British journal of surgery”. 7 (74), s. 594–596, 1987. PMID: 3304519.
- ↑ Mayo C.H. Paroxysmal hypertension with tumor of retroperitoneal nerve: report of a case. „JAMA”. 89, s. 1047–1050, 1927.
- ↑ Pincoffs M.C. A case of Paroxysmal hypertension associated with suprarenal tumor. „Trans Assoc Am Physicians”. 44, s. 295–299, 1929.
- ↑ Rabin C.B. Chromaffin cell tumor of the suprarenal medulla (pheochromocytoma). „Arch Pathol”. 7, s. 228–243, 1929. PMID.
- ↑ H.L. Mason, C.S. Myers, E.C. Kendall. The chemistry of crystalline substances isolated from the suprarenal gland. „J. Biol. Chem.”. 114, s. 613–631, 1936.
- ↑ Hyman A, Mencher W.H. Pheochromocytoma of adrenal gland. „J. Urol”. 49, s. 755–771, 1943.
- ↑ Roth G.M, Kvale W.F. A tentative test for pheochromocytoma. „Am J Med Sci”. 210, s. 653–660, 1945.
- ↑ Engel A., von Euler U.S. Diagnostic value of increased urinary output of pheochromocytoma. „Lancet”. 6630 (2), s. 387, 1950. PMID: 14785169.
- ↑ WEIL-MALHERBE H. Phaeochromocytoma catechols in urine and tumour tissue. „Lancet”. 6937 (271), s. 282–284, 1956. PMID: 13347128.
- ↑ Armstrong M.D., McMillan A., Shaw K.N. 3-Methoxy-4-hydroxy-D-mandelic acid, a urinary metabolite of norepinephrine. „Biochimica et biophysica acta”. 2 (25), s. 422–423, 1957. PMID: 13471587.
- ↑ Sipple J.H. The association of pheochromocytoma with carcinoma of the thyroid gland. „Am J Med”. 31, s. 163–166, 1961.
- ↑ Lawrence A.M. Glucagon provocative test for pheochromocytoma. „Annals of internal medicine”. 6 (66), s. 1091–1096, 1967. PMID: 6027944.
- ↑ Stewart B.H., Bravo E.L., Haaga J., Meaney T.F., Tarazi R. Localization of pheochromocytoma by computed tomography. „The New England journal of medicine”. 9 (299), s. 460–461, 1978. PMID: 683279.
- ↑ Sisson J.C., Frager M.S., Valk T.W., Gross M.D., Swanson D.P., Wieland D.M., Tobes M.C., Beierwaltes W.H., Thompson N.W. Scintigraphic localization of pheochromocytoma. „The New England journal of medicine”. 1 (305), s. 12–17, 1981. PMID: 7231514.
- ↑ Neumann H.P., Berger D.P., Sigmund G., Blum U., Schmidt D., Parmer R.J., Volk B., Kirste G. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. „The New England journal of medicine”. 21 (329), s. 1531–1538, 1993. PMID: 8105382.
Bibliografia
- Andrzej Szczeklik: Choroby wewnętrzne. Kraków: Medycyna Praktyczna, 2005, s. 1126–1128. ISBN 83-7430-031-0.
- Eisenhofer G., Bornstein S.R., Brouwers F.M., Cheung N.K., Dahia P.L., de Krijger R.R., Giordano T.J., Greene L.A., Goldstein D.S., Lehnert H., Manger W.M., Maris J.M., Neumann H.P., Pacak K., Shulkin B.L., Smith D.I., Tischler A.S., Young W.F. Malignant pheochromocytoma: current status and initiatives for future progress. „Endocrine-related cancer”. 3 (11), s. 423–436, 2004. DOI: 10.1677/erc.1.00829. PMID: 15369446.
- Pacak K. Preoperative management of the pheochromocytoma patient. „The Journal of clinical endocrinology and metabolism”. 11 (92), s. 4069–4079, 2007. DOI: 10.1210/jc.2007-1720. PMID: 17989126.
- Włodzimierz Januszewicz, Bożenna Wocial, Marek Sznajderman, Andrzej Januszewicz: Guz chromochłonny. Wyd. II. Warszawa: Wydawnictwo Lekarskie PZWL, 2000. ISBN 83-200-2344-0.
- V. Kumar, R.S. Cotran, S.L. Robbins Patologia Urban & Partner, Wrocław 2005, ISBN 0-7216-9274-5.
- Grupa Robocza PTNT. Diagnostyka i leczenie guza chromochłonnego. Zalecenia Polskiego Towarzystwa Nadciśnienia Tętniczego (1998). „Medycyna Praktyczna” 5, 1998.
Linki zewnętrzne
- Pheochromocytoma RESearch Support ORganization (PRESSOR) (ang.)
- Guz chromochłonny w eMedicine (ang.)
- Guz chromochłonny na stronie National Cancer Institute (ang.)
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.