Epigenetyka – nauka zajmująca się badaniem zmian ekspresji genów, które nie są związane ze zmianami w sekwencji nukleotydów w DNA. Ekspresja ta może być modyfikowana przez czynniki zewnętrzne i podlegać dziedziczeniu[1].
Wyraz epigenetyka składa się ze słów epi – poza czymś, w dodatku do, oraz genetyka[2]. Wymyślenie tego terminu przypisuje się Conradowi H. Waddingtonowi, który w swojej pracy z 1939 użył terminu „epigenetyczny” tłumacząc, dlaczego komórki embrionalne różnicują się w zupełnie odmienne tkanki, mimo że posiadają identyczny materiał genetyczny[3].
Epigenetyka a genetyka
Choć wszystkie komórki organizmu wielokomórkowego zawierają praktycznie identyczną informację genetyczną, identyczny genotyp, to jednak komórki różnych tkanek znacznie się różnią, wykazują odmienny fenotyp. Ta różnorodność wynika z różnego wykorzystania tej samej informacji genetycznej, dzięki czemu w przypadku organizmu człowieka powstaje około 200 różnych typów komórek. To właśnie mechanizmy epigenetyczne umożliwiają powstanie stałych wzorców ekspresji genów dla danej zróżnicowanej komórki, a jednocześnie umożliwiają krótkotrwałe, odwracalne zmiany w ich ekspresji[4]. Dzięki nim inicjowane są i utrzymywane procesy związane z różnicowaniem komórek[2].
U ssaków ważne procesy reprogramowania epigenetycznego zachodzą na etapie gametogenezy oraz przed implantacją zarodka[5]. W rozwoju osobniczym następują rozmaite zmiany w ekspresji genów w wyniku przeprogramowań epigenetycznych, które wpływają na fenotyp[6]. Gromadzone są one przez całe życie[7] i wraz z uszkodzeniami komórkowymi oraz zmniejszoną zdolnością do ich naprawy są jednym ze zjawisk kojarzonych ze starzeniem się[6].
Konsekwencje różnic epigenetycznych
Epigenomem określa się zestaw modyfikacji DNA i białek histonowych, które regulują strukturę chromatyny, a przez to kontrolują ekspresję genów, replikację i naprawę DNA oraz inne funkcje komórkowe[8]. Epigenom może sterować potencjałem genomu, określać, które geny będą aktywne, programować funkcje komórek, rozwój[9]. Różnice epigenetyczne mogą tłumaczyć różnice między fenotypami bliźniąt jednojajowych mimo takiego samego genomu. Rozbieżności te są większe, jeśli bliźnięta były wychowywane w różnych środowiskach[10]. Co więcej, epigenetyka pogłębia również różnice między zupełnie odmiennymi gatunkami mimo dużych podobieństw w genomie[9]. Przykładowo prawie każdy gen człowieka ma swój odpowiednik w genomie myszy (tzn. nie pojawiły się nowe geny od czasu wspólnego przodka 50 milionów lat temu)[11].
Narzędziami epigenomu są tzw. przełączniki (mechanizmy) epigenetyczne[12]. Działanie mechanizmów epigenetycznych jest bardziej stałe niż działanie represorów, naznaczenie może przetrwać replikację i podział komórki[13]. Wykazują one także dużą elastyczność – przeprogramowanie komórek może potencjalnie być wywołane przez takie czynniki jak m.in. wychowanie, pożywienie, stres, traumy, klimat[12]. Przykładowo u pszczół królowa i robotnice mogą być identyczne pod względem genetycznym, a jednak zupełnie różne pod względem morfologicznym. Aby wyhodować królową, larwa karmiona jest mleczkiem pszczelim przez cały okres stadium larwalnego oraz dalej w okresie dorosłym, podczas gdy larwy robotnic tylko w początkowym okresie stadium larwalnego (potem odżywiane są pyłkiem i nektarem). Odmienna dieta wystarczy dla przeprogramowania epigenetycznego, które pozwala królowej na pełne wykształcenie jajników i odmienną ekspresję ponad jednej piątej genów zarejestrowaną w komórkach mózgowych w porównaniu z komórkami robotnic[14].
Innym przykładem są niektóre zwierzęta (np. wiele gadów jak krokodyle), u których o płci przyszłego zwierzęcia decyduje temperatura otoczenia podczas wrażliwej fazy inkubacji jaja[15], a mechanizmem odpowiedzialnym za to zjawisko może być epigenetyka[16]. Również na organizmy ludzi środowisko może wpływać znacznie silniej niż się to przypuszcza[17]. Ponadto sugeruje się, że w tworzeniu różnic w rozwoju mózgu pod wpływem hormonów płciowych u kobiet i mężczyzn, w powstawaniu różnic w zachowaniu mogą pośredniczyć mechanizmy epigenetyczne[18].
Ze względu na to, że niewiele wiadomo o tym, jakie czynniki mogą wywoływać przeprogramowanie epigenetyczne, ani jakie może mieć ono konsekwencje, rośnie liczba badań w tym kierunku. Niektórzy naukowcy jak Rudolf Jaenisch czy Thomas Jenuwein twierdzą nawet, że nastał czas „postgenomicznego” myślenia w biologii[19].
Mechanizmy epigenetyczne

Rozwinięty DNA jest bardzo długi i musi zostać silnie skondensowany, by zmieścić się w strukturach komórki. U eukariotów DNA jest upakowany w niewielkim jądrze komórkowym dzięki tworzeniu kompleksów z białkami histonowymi i tworzeniu chromatyny, której podstawową jednostką strukturalną jest nukleosom. Zbudowany jest on z oktameru histonów H2A, H2B, H3 i H4, na który nawinięty jest fragment helisy DNA. Między nukleosomami występuje łącznikowy DNA. Rozluźniony sznur nukleosomów jest łatwo dostępny dla aparatu transkrypcyjnego (czynników transkrypcyjnych, polimerazy RNA). Chromatyna w takim stanie nazywana jest euchromatyną. Ekspresja genów w takich warunkach jest wzmożona. Kiedy chromatyna jest ściśle upakowana, staje się ona nieaktywna transkrypcyjnie, a ekspresja genów zahamowana. Chromatynę w takim stanie nazywa się heterochromatyną[20]. Stan heterochromatyny może utrzymywać się przez wiele generacji komórek, będąc przekazywany przy podziałach komórkowych. Na stan chromatyny wpływają zarówno modyfikacje histonów, jak i metylacja DNA[21].
Modyfikacje histonów
Białka histonowe podlegają różnym modyfikacjom. Najlepiej zbadaną modyfikacją jest mechanizm acetylacji histonów. Acetylacja histonów polega na przyłączeniu grupy acetylowej do reszty lizyny na N-końcu histonu rdzeniowego. Końce te tworzą wystające ogony, a ich acetylacja zmniejsza powinowactwo histonów do DNA, rozluźniając strukturę chromatyny. W heterochromatynie histony są przeważnie nieacetylowane. W acetylacji biorą udział acetylotransferazy histonowe (HAT). Proces odwrotny – deacetylację katalizują deacetylazy histonów (HDAC)[22].
Do innych modyfikacji histonów należy[22]:
- metylacja reszt lizynowych i argininowych na N-końcach histonów H3 i H4;
- fosforylacja reszt serynowych na N-końcach histonów H2A, H2B, H3 i H4;
- ubikwitynacja reszt lizynowych na C-końcu histonów H2A i H2B.
Do badania modyfikacji histonów można stosować techniki immunoprecypitacji chromatyny (ChIP)[23]. Badania dotyczące różnorodności modyfikacji histonów, interakcji różnych modyfikacji doprowadziła do sformułowania hipotezy o kodzie histonowym. Zakłada ona, że wzór modyfikacji histonów określa, który region genomu w danym czasie ulega ekspresji i kieruje takimi aspektami jak regulacja genomu z cyklem komórkowym czy naprawa uszkodzonych miejsc[22].
Metylacja DNA
Cytozyny w DNA mogą ulegać kowalencyjnej metylacji. Reakcję tę katalizują metylotransferazy DNA (DNMT)[24]. Wysoki poziom powstałej w ten sposób 5-metylocytozyny jest charakterystyczny dla nieaktywnej chromatyny[13]. Metylację cytozyny w DNA obserwuje się u bakterii, roślin, zwierząt, w tym ssaków, ale rzadko lub wcale u drożdży, nicieni, Drosophila melanogaster[25]. U kręgowców ok. 10% wszystkich cytozyn w genomie jest metylowanych, a u roślin do 30%. W przypadku zwierząt, metylacji ulegają jednak tylko cytozyny, które wchodzą w skład niektórych sekwencji 5’–CG–3’[26] określanych często jako CpG (gdzie p oznacza grupę fosforanową)[27]; w przypadku roślin są to sekwencje 5’–CNG–3’ (gdzie N oznacza dowolną zasadę azotową)[26].
Istnieją dwie rodzaje metylacji – zachowawcza i de novo. Metylacja zachowawcza odpowiada za metylacje nowo zsyntetyzowanej nici DNA po replikacji w miejscach komplementarnych do miejsc metylowanych w nici rodzicielskiej. Dzięki temu wzór metylacji może być dziedziczony po podziale komórki. Drugi rodzaj to metylacja de novo, która polega na przyłączaniu grup metylowych w całkowicie nowych miejscach, przez co zmianie ulega wzór metylacji[26].
Metylacja związana jest z hamowaniem ekspresji genów. Geny aktywne położone są w miejscach niemetylowanych[28]. W genomach kręgowców przed wieloma genami znajdują się tzw. wyspy CpG[29] (w przypadku człowieka dotyczy to ok. 60–70% genów[30]). Są to sekwencje długości ok. 1 kb o wyższej zawartości CpG w stosunku do średniej dla całego genomu[29]. Wyspy CpG przy genach metabolizmu podstawowego, które we wszystkich komórkach ulegają ekspresji, są niemetylowane. W przypadku genów, których ekspresja charakterystyczna jest tylko dla specyficznych tkanek, wyspy CpG są niemetylowane tylko w komórkach tych specyficznych tkanek; w innych zaś są metylowane i nie ulegają ekspresji[28]. Zatem komórki różnych organów, tkankek mają specyficzny wzór sekwencji metylowanego DNA[25]. Jest on przekazywany komórce potomnej, dzięki czemu posiada ona informację, które geny powinny ulegać ekspresji[28].
Bakterie metylują swoje DNA w celu ochrony przed enzymami restrykcyjnymi stanowiącymi część systemu restrykcji i modyfikacji, który chroni komórkę przed obcym DNA[28].
Do badania metylacji stosować można sekwencjonowanie DNA traktowanego wodorosiarczynem czy immunoprecypitację metylowanego DNA (MeDIP)[23].
Inne mechanizmy epigenetyczne
Do mechanizmów epigenetycznych zalicza się też wyciszanie genów oparte na miRNA[31] czy działanie prionów[5].
miRNA
MikroRNA (miRNA) stanowią krótkie cząsteczki RNA, które pełnią rolę regulatorów ekspresji genów[32]. miRNA łączy się z mRNA wykazującym do niego komplementarność, uniemożliwiając translację mRNA na białko (wyciszając gen)[33]. Stanowią część epigenetycznej maszynerii, jednocześnie same podlegają wpływom epigenetycznych modyfikacji podobnie jak geny kodujące białka. miRNA z jednej strony może wpływać na epigenetyczne regulatory takie jak metylotranferazy DNA, deacetylazy histonowe, białka z grupy Polycomb. Z drugiej strony około połowa genów miRNA powiązana jest w wyspami CpG. Ekspresja tych genów jest zatem zależna od metylacji DNA; ponadto wpływają na nią modyfikacje histonów[32].
Priony
Priony stanowią infekcyjne formy białek, które są zdolne zmieniać konformację innych białek. Może nastąpić więc zmiana fenotypu bez zmiany w sekwencji DNA (będąc białkami, nie mają wcale materiału genetycznego), a jednocześnie przekazywana informacja strukturalna podlega dziedziczeniu. W tym sensie stanowią przykład dziedziczenia epigenetycznego[34][35].
Monoalleliczna ekspresja genów
W komórkach diploidalnych występują dwie kopie genów – tzw. allele (inaczej może być w przypadku chromosomów płci). Jeden z alleli dziedziczy się od matki, drugi od ojca i przeważnie obie kopie ulegają ekspresji. Jednak w przypadku niewielkiej grupy genów tylko jeden allel ulega transkrypcji i translacji na białko. U ssaków do przykładów monoallelicznej ekspresji genów należy wyłączenie alleliczne, inaktywacja chromosomu X i genomowe piętnowanie rodzicielskie (imprinting). Za wyciszanie odpowiednich genów odpowiedziane są mechanizmy epigenetyczne[2].
Imprinting genomowy
Imprinting genomowy polega na tym, że ekspresja piętnowanego genu następuje tylko z jednego z dwóch chromosomów rodzicielskich. Są to zwykle bardzo ważne geny rozwojowe, a utrata piętna wiąże się z pewnymi chorobami jak zespół Pradera-Williego czy zespół Angelmana[36].
Inaktywacja chromosomu X
U samic ssaków jeden z chromosomów X podczas rozwoju embrionalnego zostaje inaktywowany w celu zrównania poziomu ekspresji genów z tego chromosomu z samcami posiadającymi tylko jedną jego kopię. Taka inaktywacja zachodzi również u polisomicznych osobników, np. w zespole Klinefeltera[37]. W wyniku epigenetycznych modyfikacji chromatyny przechodzi ona w postać heterochromatyny, a w obrazie mikroskopowym w interfazie widoczna jest ona jako ciałko Barra. Inaktywacja przenoszona jest na każdą komórkę osobno, wyłączany jest chromosom X pochodzący albo od ojca, albo od matki[38], co fenotypowo może manifestować się np. charakterystycznym mozaikowym umaszczeniem kotów. Geny na inaktywowanym chromosomie X nie są jednak zupełnie nieaktywne, nawet do 25% genów znajdujących się na nim może ulegać od czasu do czasu ekspresji[38].
Wyłączenie alleliczne
Wyłączenie alleliczne pełni istotną rolę w różnicowaniu się komórek wielu organizmów. Odbywa się głównie przez rearanżację odcinków DNA, które regulują ekspresję danych genów. Przykładowo powstawanie typu płci u drożdży wiąże się z zamianą aktywnej kasety genów związanych z typem płci znajdującej się w locus ulegającym ekspresji z drugą, dotychczas wyciszoną kasetą. Ekspresja wyciszonych kaset jest blokowana przez mechanizmy epigenetyczne[39].
Ruchome elementy genetyczne a epigenetyka
W genomach bakterii, roślin i zwierząt często spotykane są ruchome elementy genetyczne[40]. W przypadku człowieka jedynie niecałe 2% materiału genetycznego stanowią geny niosące informacje o strukturze białek[4]. Tymczasem transpozony u ssaków mogą zajmować prawie połowę genomu, a u niektórych roślin wyższych nawet 90%. Są to sekwencje DNA mogące przemieszczać się w inne miejsca w genomie; zdolne są również do powielania się. Przemieszczenie się na inne miejsce może doprowadzić do przerwania ciągłości genu, a jednak rzadko powodują mutację u roślin i zwierząt[40]. Najprawdopodobniej metylacja DNA, jeden z mechanizmów epigenetycznych, wyewoluował właśnie w celu inaktywacji takich „pasożytów genomowych” jak transpozony czy retrowirusy, co przypomina bakteryjne systemy restrykcyjne. Mechanizmy rozpoznawania, metylacji i inaktywacji powtarzających się fragmentów DNA u roślin, grzybów i zwierząt prowadzą często do niepowodzeń we wprowadzaniu transgenów metodami inżynierii genetycznej[41] oraz przy klonowaniu organizmów[42].
W celu ochrony DNA komórki gospodarza przed takimi sekwencjami jak transpozony mogą w ich obrębie ulec metylacji cytozyny. Metylowana cytozyna ma tendencję do tranzycji do tyminy, powodując mutację i inaktywację transpozonów, a ponadto metylacja wycisza ekspresję tych elementów[43]. Inną linią obrony jest występowanie heterochromatyny w rejonie występowania transpozonów. Hamuje to ich transkrypcję, przemieszczanie się i aktywność rekombinacyjną[21].
Epigenetyka a zdrowie
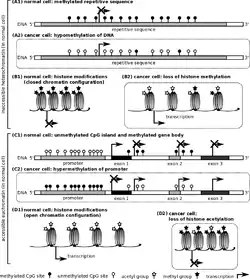
Nieprawidłowe znakowanie epigenetyczne jest również związane z wieloma chorobami. Modyfikacje epigenetyczne odgrywają kluczową rolę w wielu rodzajach nowotworów, chorobie Alzheimera i innych chorobach neurodegradacyjnych[5]. Różne zmiany epigenetyczne mogą wywierać wpływ na takie choroby jak m.in. cukrzyca[44], choroby reumatyczne[45], nadciśnienie[46], depresja, schizofrenia, uzależnienia[47].
Wiele czynników środowiskowych może być przyczyną stresu oksydacyjnego w komórce. Uszkodzone w ten sposób DNA może prowadzić do nieprawidłowego działania metylotransferaz DNA. Wpływ na metylację DNA ma np. zanieczyszczenie powietrza, stres, dym papierosowy. W konsekwencji epigenetyka stanowi niejako mechanizm pośredniczący między genetyką lub środowiskiem a chorobą. Co więcej, niektóre choroby są bezpośrednio wywołane przez pewne modyfikacje epigenetyczne. Należą do nich zaburzenia imprintingu w zespole Beckwitha-Wiedemanna. Niektóre choroby są wywołane mutacjami genów, które pociągają za sobą niekorzystne zmiany w mechanizmach epigenetycznych. Przykładowo przyczyną zespołu Retta jest mutacja w genie MECP2. Nieprawidłowe białko MECP2 zmienia ekspresję innych genów, które normalnie są regulowane przez mechanizmy epigenetyczne[5]. Innym przykładem jest zespół ICF związany z nieprawidłowym działaniem metylotransferazy odpowiedzialnej za kontrolę metylacji DNA[48].
Ponadto czynniki epigenetyczne mogą służyć jako biomarkery do wykrywania chorób. Przykładowo gen S-transferazy glutationowej (GSTP1) jest hipermetylowany w przypadku raka prostaty i połączenie standardowego testu PSA z diagnostyką wykorzystującą epigenetyczne biomarkery pozwala zmniejszyć liczbę fałszywych wyników dodatnich[5].
W komórkach rakowych spotyka się nadmierną metylację wysp CpG, co może prowadzić do wyciszenia genów supresorowych, i niski poziom metylacji reszty genomu, co może zwiększać ekspresję onkogenów[49][50]. Demetylacja w skali genomu postępuje wraz z wiekiem, najczęściej jej efektem jest niestabilność chromosomów[49]. Poza tym w wielu rodzajach komórek nowotworowych obserwuje się brak grupy acetylowej w lizynie 16 i trimetylowej w lizynie 20 histonu H4[49][50]. W celu przywrócenia ekspresji genów supresorowych wyciszonych przez mechanizmy epigenetyczne stosuje się czasem terapię z zastosowaniem inhibitorów metylotransferaz DNA lub inhibitorów deacetylaz histonowych. Do lecznictwa zostały zatwierdzone leki takie jak Vorinostat i romidepsyna[51].
Przypisy
- ↑ Smith D.: Epigenetics. W: Kreutzer J.: Encyclopedia of Clinical Neuropsychology. Springer, 2017. DOI: 10.1007/978-3-319-56782-2_9029-1. ISBN 978-3-319-56782-2.
- 1 2 3 Allison 2007 ↓, s. 393.
- ↑ Popova E., Barnstable C. J.. Epigenetics Rules. „Journal of Ocular Biology, Diseases, and Informatics”. 4, s. 93–94, 2011. DOI: 10.1007/s12177-012-9088-8.
- 1 2 Licznerska, Ignatowicz i Baer-Dubowska 2012 ↓, s. 60–61.
- 1 2 3 4 5 Bakulski K. M., Fallin M. D.. Epigenetic Epidemiology: Promises for Public Health Research. „Environmental and MolecularMutagenesis”. 55, s. 171–183, 2014. DOI: 10.1002/em.21850.
- 1 2 Jones M. J., Goodman S. J., Kobor M. S.. DNA Methylation and Healthy Human Aging. „Aging Cell”. 14, s. 924–932, 2015. DOI: 10.1111/acel.12349.
- ↑ Spork 2011 ↓, s. 110.
- ↑ McCullough S. D., Dhingra R., Fortin M. C., Diaz-Sanchez D.. Air Pollution and the Epigenome: A Model Relationship for the Exploration of Toxicoepigenetics. „Current Opinion in Toxicology”. 6, s. 18–25, 2017. DOI: 10.1016/j.cotox.2017.07.001.
- 1 2 Spork 2011 ↓, s. 11.
- ↑ Licznerska, Ignatowicz i Baer-Dubowska 2012 ↓, s. 76.
- ↑ Watson J. D., Baker T. A., Bell S. P., Gann A., Levine M., Losick R.: Molecular Biology of the Gene. Pearson Education, 2004, s. 614. ISBN 0-321-22368-3.
- 1 2 Spork 2011 ↓, s. 12–13.
- 1 2 Fletcher, Hickey i Winter 2010 ↓, s. 77.
- ↑ Chittka A., Chittka L.. Epigenetics of Royality. „PLoS Biology”. 8 (11), 2010. DOI: 10.1371/journal.pbio.1000532.
- ↑ Spork 2011 ↓, s. 91–92.
- ↑ Shen Z. G., Wang H. P.. Molecular Players Involved in Temperature-dependent Sex Determination and Sex Differentiation in Teleost Fish. „Genetics Selection Evolution”. 46 (1), s. 26, 2014. DOI: 10.1186/1297-9686-46-26.
- ↑ Spork 2011 ↓, s. 94.
- ↑ McCarthy M. M., Nugent B. M.. Epigenetic Contributions to Hormonally-Mediated Sexual Differentiation of the Brain. „Journal of Neuroendocrinology”. 25, s. 1133–1140, 2013. DOI: 10.1111/jne.12072.
- ↑ Spork 2011 ↓, s. 15.
- ↑ Licznerska, Ignatowicz i Baer-Dubowska 2012 ↓, s. 61–62.
- 1 2 Allison 2007 ↓, s. 425.
- 1 2 3 Brown 2013 ↓, s. 280–283.
- 1 2 Stolerman I. P.: Encyclopedia of Psychopharmacology. Springer, 2010, s. 487–489. DOI: 10.1007/978-3-540-68706-1.
- ↑ Licznerska, Ignatowicz i Baer-Dubowska 2012 ↓, s. 63.
- 1 2 Korochkin L. I.. What is Epigenetics. „Russian Journal of Genetics”. 42 (9), s. 958–965, 2006. DOI: 10.1134/S102279540609002X.
- 1 2 3 Brown 2013 ↓, s. 285.
- ↑ Allison 2007 ↓, s. 394.
- 1 2 3 4 Brown 2013 ↓, s. 286.
- 1 2 Brown 2013 ↓, s. 137.
- ↑ Illingworth R. S., Bird A. P.. CpG Islands – ‘A Rough Guide’. „FEBS Letters”. 583, s. 1713–1720, 2009. DOI: 10.1016/j.febslet.2009.04.012.
- ↑ Charon K. M., Świtoński M.: Genetyka i Genomika Zwierząt. Warszawa: Wydawnictwo Naukowe PWN, 2012, s. 71. ISBN 978-83-01-17107-0.
- 1 2 Piletič K., Kunej T.. MicroRNA Epigenetic Signatures in Human Disease. „Archives of Toxicology”. 90, s. 2405–2419, 2016. DOI: 10.1007/s00204-016-1815-7.
- ↑ Loginov V. I., Rykov S. V., Fridman M. V., Braga E. A.. Methylation of miRNA Genes and Oncogenesis. „Biochemistry (Moscow)”. 80 (2), s. 145–162, 2015. DOI: 10.1134/S0006297915020029.
- ↑ Yool A., Edmunds W. J.. Epigenetic Inheritance and Prions. „Journal of Evolutionary Biology”. 11, s. 241–242, 1998.
- ↑ Manjrekar J.. Epigenetic Inheritance, Prions and Evolution. „Journal of Genetics”. 96 (3), s. 445–456, 2017. DOI: 10.1007/s12041-017-0798-3.
- ↑ Allison 2007 ↓, s. 398–399.
- ↑ Fletcher, Hickey i Winter 2010 ↓, s. 79.
- 1 2 Allison 2007 ↓, s. 410–412.
- ↑ Allison 2007 ↓, s. 426–428.
- 1 2 Allison 2007 ↓, s. 412–413.
- ↑ Fletcher, Hickey i Winter 2010 ↓, s. 83.
- ↑ Jablonka E., Lamb M. J.. The Changing Concept of Epigenetics. „Annals of the New York Academy of Sciences”. 981, s. 82–96, 2002.
- ↑ Allison 2007 ↓, s. 423.
- ↑ Al-Haddad R., Karnib N., Assaad R. A., Bilen Y., Emmanuel N., Ghanem A., Younes J., Zibara V., Stephan J. S., Sleiman S. F.. Epigenetic Changes in Diabetes. „Neuroscience Letters”. 625, s. 64–69, 2016. DOI: 10.1016/j.neulet.2016.04.046.
- ↑ Husain A., Jeffries M. A.. Epigenetics and Bone Remodeling. „Current Osteoporosis Reports”, 2017. DOI: 10.1007/s11914-017-0391-y.
- ↑ Millis R. M.. Epigenetics and Hypertension. „Current Hypertension Reports”. 13, s. 21–28, 2011. DOI: 10.1007/s11906-010-0173-8.
- ↑ Mahgoub M., Monteggia L. M.. Epigenetics and Psychiatry. „Neurotherapeutics”. 10, s. 734–741, 2013. DOI: 10.1007/s13311-013-0213-6.
- ↑ Fletcher, Hickey i Winter 2010 ↓, s. 82.
- 1 2 3 Allison 2007 ↓, s. 396.
- 1 2 Glozak M. A., Seto E.. Histone Deacetylases and Cancer. „Oncogene”. 26, s. 5420–5432, 2007. DOI: 10.1038/sj.onc.1210610.
- ↑ Licznerska, Ignatowicz i Baer-Dubowska 2012 ↓, s. 72.
Bibliografia
- T. A. Brown: Genomy. Warszawa: Wydawnictwo Naukowe PWN, 2013. ISBN 978-83-01-15634-3.
- B. Licznerska, E. Ignatowicz, W. Baer-Dubowska: Biologia Molekularna dla Farmaceutów. Poznań: Wydawnictwo Naukowe Uniwersytetu Medycznego im. Karola Marcinkowskiego w Poznaniu, 2012. ISBN 978-83-7597-146-0.
- L. A. Allison: Podstawy Biologii Molekularnej. Warszawa: Wydawnictwa Uniwersytetu Warszawskiego, 2007. ISBN 978-83-235-0527-3.
- H. Fletcher, I. Hickey, P. Winter: Krótkie Wykłady: Genetyka. Warszawa: Wydawnictwo Naukowe PWN, 2010. ISBN 978-83-01-16343-3.
- P. Spork: Drugi kod. Epigenetyka, czyli jak Możemy Sterować Własnymi Genotypami. Warszawa: Wydawnictwa W.A.B., 2011. ISBN 978-83-7747-542-3.