| Paralysis supranuclearis progressiva | |
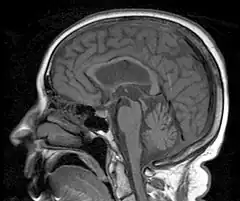 Obraz MR T1-zależny pokazuje atrofię śródmózgowia z zachowaniem objętości mostu i zanik pokrywy śródmózgowia, zwłaszcza wzgórków górnych. Obraz ten razem z objawami klinicznymi (postępująca demencja, ataksja, inkontynencja, oczopląs) sugeruje rozpoznanie postępującego porażenia nadjądrowego | |
| Klasyfikacje | |
| ICD-10 | |
|---|---|
| DiseasesDB | |
| OMIM | |
| MeSH | |
Postępujące porażenie nadjądrowe (choroba/zespół Steele’a-Richardsona-Olszewskiego, łac. paralysis supranuclearis progressiva[1] ang. progressive supranuclear palsy, PSP, Steele-Richardson-Olszewski syndrome) – rzadka choroba neurodegeneracyjna z grupy tauopatii, objawiająca się zespołem parkinsonowskim i otępieniem.
Epidemiologia
Choroba określana jest jako rzadka; jej częstość szacuje się na 6,5:100 000[2]. Występuje w starszym wieku, najczęściej w 6. dekadzie życia.
Etiologia
Ze względu na etiologię PSP zaliczane jest do tauopatii. W mózgowiu obserwuje się postępujący zanik, glejozę i zwyrodnienie neurofibrylarne komórek nakrywki śródmózgowia, istoty czarnej i czasem kory płatów skroniowych. W związku z zanikami dochodzi do poszerzenia III i IV komory mózgu.
Objawy i przebieg
Na obraz zespołu składają się:
- symetryczny zespół parkinsonowski ze sztywnością mięśniową (osiową) i bradykinezją (spowolnieniem ruchowym), częstymi upadkami
- zespół rzekomoopuszkowy
- zaburzenia ruchomości gałek ocznych; początkowo dotyczy kierunku pionowego (objaw Parinauda), potem wszystkich kierunków
- dystoniczne odchylenie głowy ku tyłowi (retrocollitis)
- otępienie podkorowe ze spowolnieniem intelektualnym (bradyfrenia), dysfazją, trudnościami w wykonywaniu złożonych czynności
- zaburzenia psychiczne, zmiany osobowości (apatia, agresja, perseweracje, zaburzenia seksualne).
Charakterystycznym objawem jest dodatni tzw. objaw klaskania (ang. applause sign). Pacjent proszony jest o szybkie zaklaskanie w ręce trzy razy; pacjenci z PSP wykonują więcej klaśnięć. W jednym badaniu test pozwolił na zróżnicowanie PSP z chorobą Parkinsona (p < 0,00) i otępieniem czołowo-skroniowym (p < 0,001)[3].
Choroba nieodwracalnie prowadzi do całkowitego unieruchomienia. Mediana przeżycia wynosi mniej niż 6 lat od momentu postawienia rozpoznania.
Rozpoznanie
Zaproponowano kryteria rozpoznania PSP. „Prawdopodobne” rozpoznanie można postawić, gdy spełnione są przynajmniej trzy kryteria dodatkowe, „możliwe” gdy spełnione są dwa kryteria dodatkowe + kryteria podstawowe[4].
- Kryteria podstawowe
- wiek powyżej 40 lat
- postępujący przebieg
- oftalmoplegia nadjądrowa
- Kryteria dodatkowe
Rozpoznanie różnicowe
W diagnostyce różnicowej PSP należy uwzględnić przede wszystkim chorobę Parkinsona we wczesnym stadium, także inne choroby neurodegeneracyjne z otępieniem i parkinsonizmem (np. otępienie z ciałami Lewy’ego). Z chorobą Parkinsona PSP różnicuje słaba odpowiedź na lewodopę i symetryczność objawów parkinsonowskich[5].
Leczenie
Nie jest znane skuteczne leczenie. Próby stosowania lewodopy, agonistów amantadyny i dopaminy są mało skuteczne. Postęp choroby prowadzi nieodwracalnie do unieruchomienia chorego i konieczności żywienia pozajelitowego. Przyczyną śmierci są powikłania związane z unieruchomieniem pacjenta.
Historia
Chorobę opisali Jerzy Olszewski, John Clifford Richardson i John Steele w 1963 roku[6][7].
Przypisy
- ↑ Andrzej Szczeklik, Piotr Gajewski: Interna Szczeklika 2017. Wydawnictwo Medycyna Praktyczna, 2017. ISBN 978-83-7430-517-4.
- ↑ J.C. Steele. Progressive supranuclear palsy. Historical notes.. „J Neural Transm Suppl”. 42, s. 3-14, 1994. PMID: 7964694.
- ↑ B. Dubois, A. Slachevsky, B. Pillon, R. Beato i inni. "Applause sign" helps to discriminate PSP from FTD and PD.. „Neurology”. 64 (12), s. 2132-2133, 2005. DOI: 10.1212/01.WNL.0000165977.38272.15. PMID: 15985587.
- ↑ Neurologia. Podręcznik dla studentów medycyny. Wojciech Kozubski, Paweł P. Liberski (red.). Warszawa: Wydawnictwo Lekarskie PZWL, 2006, s. 286-287. ISBN 83-200-3244-X.
- ↑ I. Litvan, G. Campbell, CA. Mangone, M. Verny i inni. Which clinical features differentiate progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) from related disorders? A clinicopathological study. „Brain”. 120 ( Pt 1), s. 65-74, 1997. PMID: 9055798.
- ↑ J.C. Richardson, J. Steele, J. Olszewski. Supranuclear ophthalmoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight cases of „heterogeneous system degeneration”. „Transactions of the American Neurological Association”. 88, s. 25-29, 1963. PMID: 14272249.
- ↑ J.C. Steele, J.C. Richardson, J. Olszewski. Progressive supranuclear palsy: a heterogeneous degeneration involving brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. „Arch Neurol”. 10, s. 333-359, 1964. DOI: 10.1001/archneur.1964.00460160003001. PMID: 14107684.
Linki zewnętrzne
- SUPRANUCLEAR PALSY, PROGRESSIVE, 1; PSNP1 w bazie Online Mendelian Inheritance in Man (ang.)
- Steele-Richardson-Olszewski disease w bazie Who Named It (ang.)
- Strona PSP Association. pspeur.org. [zarchiwizowane z tego adresu (2008-05-11)].
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.