| Klasyfikacje | |
| ICD-10 | |
|---|---|
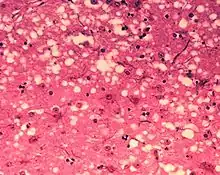
Pasażowalne encefalopatie gąbczaste (ang. Transmissible spongiform encephalopathies, TSE), choroby prionowe – choroby układu nerwowego ludzi i zwierząt spowodowane nagromadzeniem nieprawidłowo pofałdowanych białek zwanych prionami.
Etiopatogeneza i postacie
Przyczyną zakaźnych encefalopatii gąbczastych są priony – zmienione białka powszechnie występujące w komórkach nerwowych, różniące się od ich fizjologicznych odpowiedników tylko niewłaściwą strukturą przestrzenną. Białka takie mogą powstawać na skutek somatycznej (więc niedziedzicznej) mutacji genu je kodującego, a także bez występowania mutacji wskutek powolnego, samorzutnego przekształcania się białek prawidłowych w nieprawidłowe lub też na skutek odziedziczenia wadliwego genu. Ponadto w pewnych warunkach wszczepienie zdrowemu człowiekowi lub zwierzęciu białka prionowego (od chorego zwierzęcia lub człowieka) powoduje narastanie ilości zmienionego białka i w rezultacie pojawienie się objawów encefalopatii gąbczastej. Pozwala to podzielić choroby prionowe na sporadyczne, rodzinne i przepasażowane.
| Ważniejsze choroby prionowe człowieka | ||
| Choroba sporadyczna | Choroba rodzinna | Choroba przepasażowana |
| Choroba Creutzfeldta-Jakoba, postać sporadyczna (sCJD) (ok. 85–90% przypadków CJD) |
Choroba Creutzfeldta-Jakoba, postać rodzinna (fCJD), w tym zespół Gerstmanna-Sträusslera-Scheinkera (GSS) (ok. 10–15% przypadków CJD) |
Choroba Creutzfeldta-Jakoba, postacie przepasażowane – jatrogenna (iCJD) i nowy wariant (vCJD) |
| Śmiertelna bezsenność – przypadki spontaniczne (sFI) | Śmiertelna rodzinna bezsenność (FFI) | |
| Kuru (nie występuje) | ||
Prawidłowe białko prionowe, określane jako PrP-C (ang. cellular prion-related protein – komórkowe białko powiązane z prionem) ma masę 30kD i występuje w błonach komórkowych wielu tkanek, w tym bardzo obficie w błonie komórek nerwowych. Jego fizjologiczna funkcja nie została jak do tej pory poznana. Białko to kodowane jest przez gen PRNP zlokalizowany na 20 chromosomie. Białko patologiczne – PrP-Sc[1] (od scrapie, pierwszej opisanej choroby prionowej), w przeciwieństwie do PrP-C jest nierozpuszczalne w wodzie oraz oporne na działanie proteaz, czyli enzymów trawiących białka. Fizjologiczne białko prionowe w swej strukturze przestrzennej posiada trzy α-helisy i dwie tzw. β-harmonijki, przypuszcza się, że w trakcie przekształcania PrP-C w PrP-Sc następuje zmiana właśnie struktury przestrzennej dając w rezultacie białko bogate w beta-harmonijki. Przebieg tego zjawiska nie jest znany. PrP-Sc mają możliwość wymuszania zmiany konformacji przestrzennej białek PrP-C, mechanizm tego zjawiska jest jednak również niewiadomy. W patologii znaczenie ma postępujący, podostry przebieg „zakażeń” – z biegiem czasu wzrasta ilość białka PrP-Sc, stąd nie dziwi że choroby prionowe są chorobami najczęściej osobników starych.
Pojawienie się białek nieprawidłowych prawdopodobnie nie stymuluje jakiejkolwiek odpowiedzi immunologicznej.
Postacie wrodzone występują szczególnie często w niektórych nacjach – u Żydów z Libanu zapadalność jest 30-krotnie większa niż przeciętna.
Postacie przepasażowane u ludzi występują bardzo rzadko. Obecnie związane mogą być albo z działalnością medyczną (głównie: leczenie hormonem wzrostu lub gonadotropiną uzyskaną z ludzkich przysadek, implantacje opony twardej, rogówki), albo z nowym wariantem CJD gdzie dochodzi do przeniesienia zakażenia ze zwierząt (bydła z BSE) na ludzi. Najprawdopodobniej historycznym przykładem jest kuru, choroba występująca u nowogwinejskiego plemienia praktykującego rytualny kanibalizm.
Historia
Pierwszą chorobą prionową była scrapie (od ang. „drapać”) w Polsce nazywana chorobą kłusową występująca u owiec w Islandii, zaś pierwszą wykrytą TSE człowieka była kuru (inaczej curu, od „drżeć”) u plemienia kanibali z Nowej Gwinei. Odkrywcą prionów był S. Prusiner, który za swoje prace otrzymał w 1997 roku nagrodę Nobla z dziedziny medycyny.
Przykłady chorób TSE i ich nazewnictwo polskie i angielskie
| Skrót | Nazwa polska | Nazwa angielska | Nazwa zwyczajowa | Gatunek, u którego choroba występuje |
|---|---|---|---|---|
| CJD | Choroba Creutzfeldta-Jakoba | Creutzfeldt-Jakob Disease | Człowiek | |
| vCJD | Nowy wariant choroby Creutzfeldta-Jakoba | variant Creutzfeldt-Jakob Disease | Człowiek | |
| Kuru | Curu, pisane także przez K | Śmiejąca się śmierć | Człowiek | |
| FFI | Śmiertelna bezsenność rodzinna | Fatal familial insomnia | Człowiek | |
| GSS | Zespół Gerstmanna-Sträusslera-Scheinkera | Gerstmann-Sträussler-Scheinker syndrome | Człowiek | |
| BSE | Encefalopatia gąbczasta bydła | Bovine spongiform encephalopathy | Choroba szalonych krów | Bydło |
| Kołowacizna owiec | Scrapie | Owca | ||
| TME | Pasażowalna encefalopatia norek | Transmissible mink encephalopathy | Norka amerykańska | |
| CWD | Przewlekła choroba wyniszczająca | Chronic wasting disease | Jeleniowate | |
| FSE | Encefalopatia gąbczasta kotów | Feline spongiform encephalopathy | Kot | |
| EUE | brak polskiej nomenklatury | Exotic ungulate encephalopathy | Niala, Kudu |
Klasyfikacja ICD10
| kod ICD10 | nazwa choroby |
|---|---|
| ICD-10: A81 | Zakażenie powolnymi wirusami ośrodkowego układu nerwowego |
| ICD-10: A81.0 | Choroba Jacoba-Creutzfeldta |
| ICD-10: A81.8 | Inne zakażenia powolnymi wirusami ośrodkowego układu nerwowego |
| ICD-10: A81.9 | Zakażenia powolnymi wirusami ośrodkowego układu nerwowego, nieokreślone |
Przypisy
- ↑ Spotkać można także inne oznaczenia tego białka: PrP-D (disease) i PrP-CJD.
Bibliografia
- Jacek Grzybowski, Piotr Zaborowski: Teoretyczne i praktyczne podstawy infekcjologii. Wyd. I. Warszawa: Instytut Problemów Ochrony Zdrowia, 2007, s. 50–51. ISBN 978-83-60955-00-0.
- Zakażenia powolne. W: Jacek Juszczyk: Choroby zakaźne i pasożytnicze. Zdzisław Dziubek (red.). Wyd. II poprawione i uzupełnione. Warszawa: Wydawnictwo Lekarskie PZWL, 2000, s. 371–374. ISBN 83-200-2393-9.
- Paweł P. Liberski: Choroba Creutzfeldta-Jakoba i inne choroby wywołane przez priony – pasażowalne encefalopatie gąbczaste. Lublin: Czelej, 2003. ISBN 83-89309-06-8.
- Choroby wywołane przez priony. W: Paweł P. Liberski: Choroby zakaźne i pasożytnicze. Zdzisław Dziubek (red.). Wyd. III uaktualnione. Warszawa: Wydawnictwo Lekarskie PZWL, 2003, s. 372–385. ISBN 83-200-2748-9.
- Choroby prionowe u człowieka. W: Jerzy Kulczycki: Choroby zakaźne i pasożytnicze. Janusz Cianciara, Jacek Juszczyk (red.). Wyd. I. Lublin: Wydawnictwo Czelej, 2007, s. 613–618. ISBN 978-83-60608-34-0.
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.