
Photochlorination is a chlorination reaction that is initiated by light. Usually a C-H bond is converted to a C-Cl bond. Photochlorination is carried out on an industrial scale. The process is exothermic and proceeds as a chain reaction initiated by the homolytic cleavage of molecular chlorine into chlorine radicals by ultraviolet radiation. Many chlorinated solvents are produced in this way.
History

Chlorination is one of the oldest known substitution reactions in chemistry. The French chemist Jean-Baptiste Dumas investigated the substitution of hydrogen for chlorine by acetic acid in candle wax as early as 1830.[1] He showed that for each mole of chlorine introduced into a hydrocarbon, one mole of hydrogen chloride is also formed and noted the light-sensitivity of this reaction.[2] The idea that these reactions might be chain reactions is attributed to Max Bodenstein (1913). He assumed that in the reaction of two molecules not only the end product of the reaction can be formed, but also unstable, reactive intermediates which can continue the chain reaction.[3]
Photochlorination garnered commercial attention with the availability of cheap chlorine from chloralkali electrolysis.[4]
Chlorinated alkanes found an initial application in pharyngeal sprays. These contained chlorinated alkanes in relatively large quantities as solvents for chloramine T from 1914 to 1918. The Sharpless Solvents Corporation commissioned the first industrial photochloration plant for the chlorination of pentane in 1929.[5] The commercial production of chlorinated paraffins for use as high-pressure additives in lubricants began around 1930.[6] Around 1935 the process was technically stable and commercially successful.[5] However, it was only in the years after World War II that a greater build-up of photochloration capacity began. In 1950, the United States produced more than 800,000 tons of chlorinated paraffin hydrocarbons. The major products were ethyl chloride, tetrachlorocarbon and dichloromethane.[7] Because of concerns about health and environmentally relevant problems such as the ozone depletion behavior of light volatile chlorine compounds, the chemical industry developed alternative procedures that did not require chlorinated compounds. As a result of the following replacement of chlorinated by non-chlorinated products, worldwide production volumes have declined considerably over the years.[6][8]
Reactions
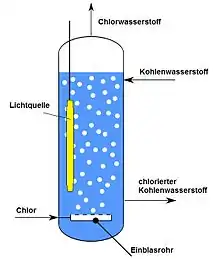
Photochlorinations are usually effected in the liquid phase, usually employing chemically inert solvents.
Alkane substrates
The photochlorination of hydrocarbon is unselective, although the reactivity of the C-H bonds is tertiary>secondary>primary. At 30 °C the relative reaction rates of primary, secondary and tertiary hydrogen atoms are in a relative ratio of approximately 1 to 3.25 to 4.43. The C-C bonds remain unaffected.[9] [10]
Upon radiation the reaction involves alkyl and chlorine radicals following a chain reaction according to the given scheme:





Chain termination occurs by recombination of chlorine atoms.[11] Impurities such as oxygen (present in electrochemically obtained chlorine) also cause chain termination.
The selectivity of photochlorination (with regard to substitution of primary, secondary or tertiary hydrogens) can be controlled by the interaction of the chlorine radical with the solvent, such as benzene, tert-butylbenzene or carbon disulfide.[12] Selectivity increases in aromatic solvents.[13] By varying the solvent the ratio of primary to secondary hydrogens can be tailored to ratios between 1: 3 to 1: 31.[14] At higher temperatures, the reaction rates of primary, secondary and tertiary hydrogen atoms equalize. Therefore, photochlorination is usually carried out at lower temperatures.[9]
Aromatic substrates
The photochlorination of benzene proceeds also via a radical chain reaction:[15]


- […]

In some applications, the reaction is carried out at 15 to 20 °C. At a conversion of 12 to 15% the reaction is stopped and the reaction mixture is worked up.[15]
Products
Chloromethanes

An example of photochlorination at low temperatures and under ambient pressure is the chlorination of chloromethane to dichloromethane. The liquefied chloromethane (boiling point -24 °C) is mixed with chlorine in the dark and then irradiated with a mercury-vapor lamp. The resulting dichloromethane has a boiling point of 41 °C and is later separated by distillation from methyl chloride.[16]
The photochlorination of methane has a lower quantum yield than the chlorination of dichloromethane. Due to the high light intensity required, the intermediate products are directly chlorinated, so that mainly tetrachloromethane is formed.[16]
Chlorinated paraffins
A major application of photochlorination is the production of chloroparaffins. Mixtures of complex composition consisting of several chlorinated paraffins are formed. Chlorinated paraffins have the general sum formula CxH(2x−y+2)Cly and are categorized into three groups: Low molecular weight chlorinated paraffins are short chain chloroparaffins (SCCP) with 10 to 13 carbon atoms, followed by medium chain chloroparaffins (MCCP) with carbon chain lengths of 14 to 17 carbon atoms and long chain chlorinated paraffins (LCCP), owing a carbon chainwith more than 17 carbon atoms. Approximately 70% of the chloroparaffins produced are MCCPs with a degree of chlorination from 45 to 52%. The remaining 30% are divided equally between SCCP and LCCP.[6] Short chain chloroparaffins have high toxicity and easily accumulate in the environment. The European Union has classified SCCP as a category III carcinogen and restricted its use.[17]
In 1985 the world production was 300,000 tonnes; since then the production volumes are falling in Europe and North America.[18] In China, on the other hand, production rose sharply. China produced more than 600,000 tonnes of chlorinated paraffins in 2007, while in 2004 it was less than 100,000 tonnes.[19]
The quantum yield for the photochlorination of n-heptane is about 7000, for example.[20] In photochlorination plants, the quantum yield is about 100. In contrast to the thermal chlorination, which can utilize the formed reaction energy, the energy required to maintain the photochemical reaction must be constantly delivered.[21]
The presence of inhibitors, such as oxygen or nitrogen oxides, must be avoided. Too high chlorine concentrations lead to high absorption near the light source and have a disadvantageous effect.[14]
Benzyl chloride, benzal chloride and benzotrichloride
The photochlorination of toluene is selective for the methyl group. Mono- to trichlorinated products are obtained. The most important of which is the mono-substituted benzyl chloride, which is hydrolyzed to benzyl alcohol. Benzyl chloride can also be converted via benzyl cyanide with subsequent hydrolysis into phenylacetic acid.[22][23] The disubstituted benzal chloride is converted to benzaldehyde, a popular flavorant[24] and intermediate for the production of malachite green and other dyes.[25] The trisubstituted benzotrichloride is used for the hydrolysis of the synthesis of benzoyl chloride:[26]


By reaction with alcohols, benzoyl chloride can be converted into the corresponding esters. With sodium peroxide it turns into dibenzoyl peroxide, a radical initiator for polymerizations. However, the atom economy of these syntheses is poor, since stoichiometric amounts of salts are obtained.
Process variants
Sulfochlorination
The sulfochlorination first described by Cortes F. Reed in 1936 proceeds under almost identical conditions as the conventional photochlorination.[27] In addition to chlorine, sulfur dioxide is also introduced into the reaction mixture. The products formed are alkylsulfonyl chlorides, which are further processed into surfactants.[28]
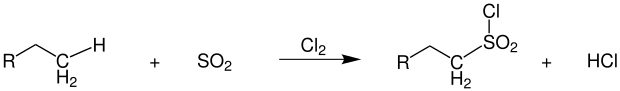
Hydrochloric acid is formed as a coupling product, as is the case with photochlorination. Since direct sulfonation of the alkanes is hardly possible, this reaction has proven to be useful. Due to chlorine, which is bound directly to the sulfur, the resulting products are highly reactive. As secondary products there are alkyl chlorides formed by pure photochlorination, as well as several sulfochlorinated products in the reaction mixture.[29]
Photobromination

Photobromination with elemental bromine proceeds analogous to photochlorination also via a radical mechanism. In the presence of oxygen, the hydrogen bromide formed is partly oxidised back to bromine, resulting in an increased yield. Because of the easier dosage of the elemental bromine and the higher selectivity of the reaction, photobromination is preferred over photochlorination at laboratory scale. For industrial applications, bromine is usually too expensive (as it is present in sea water in small quantities only and produced from oxidation with chlorine).[30][31] Instead of elemental bromine, N-bromosuccinimide is also suitable as a brominating agent.[32] The quantum yield of photobromination is usually much lower than that of photochlorination.
Further reading
- H. Hartig: Einfache Dimensionierung, photochemischer Reaktoren. In: Chemie Ingenieur Technik – CIT. 42, 1970, p. 1241–1245, doi:10.1002/cite.330422002.
- Dieter Wöhrle, Michael W. Tausch, Wolf-Dieter Stohrer: Photochemie: Konzepte, Methoden, Experimente. Wiley & Sons, 1998, ISBN 978-3-527-29545-6, p. 271–275.
- US Grant 1379367, F. Sparre & W. E. Masland, "Process of Chlorination", issued 1921-05-24, assigned to Du Pont
- US Grant 1459777, R. Leiser & F. Ziffer, "Process and Apparatus for the Chlorination of Methane", issued 1920-02-14, assigned to Ziffer Fritz and Leiser Richard
- David A. Mixon, Michael P. Bohrer, Patricia A. O’Hara: Ultrapurification of SiCl4 by photochlorination in a bubble column reactor. In: AIChE Journal. 36, 1990, p. 216–226, doi:10.1002/aic.690360207.
- Holleman, Arnold Frederik; Wiberg, Egon (2001), Wiberg, Nils (ed.), Inorganic Chemistry, translated by Eagleson, Mary; Brewer, William, San Diego/Berlin: Academic Press/De Gruyter, p. 71, ISBN 0-12-352651-5
- Hans Von Halban: Die Lichtabsorption des Chlors. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 28, 1922, p. 496–499, doi:10.1002/bbpc.19220282304.
- Arthur John Allmand: Part I.—Einstein’s law of photochemical equivalence. Introductory address to Part I. In: Trans. Faraday Soc. 21, 1926, p. 438, doi:10.1039/TF9262100438.
- Marion L. Sharrah, Geo. C. Feighner: Synthesis of Dodecylbenzen – Synthetic Detergent Intermediate. In: Industrial & Engineering Chemistry. 46, 1954, p. 248–254, doi:10.1021/ie50530a020.
- Directive 2003/53/EC of the European Parliament and of the Council of 18 June 2003 amending for the 26th time Council Directive 76/769/EEC relating to restrictions on the marketing and use of certain dangerous substances and preparations (nonylphenol, nonylphenol ethoxylate and cement)
- C. Decker, M. Balandier, J. Faure: Photochlorination of Poly(vinyl Chloride). I. Kinetics and Quantum Yield. In: Journal of Macromolecular Science: Part A – Chemistry. 16, 2006, p. 1463–1472, doi:10.1080/00222338108063248.
- Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.), Otto Bayer, Hans Meerwein, Karl Ziegler: Methoden der organischen Chemie. V/3 Fluorine and Chlorine Compounds . Thieme Verlag, Stuttgart 1962, ISBN 978-3-13-203004-6, p. 524.
- US Grant 2046090, Cortes F. Reed, "Method of halogenating compounds and product resulting therefrom", issued 1936-06-30, assigned to Charles L Horn
- R. Newe, P. Schmidt, K. Friese, B. Hösselbarth: Das Verfahren der strahlenchemischen Chlorierung von Polyvinylchlorid. In: Chemische Technik, 41(4), 1989, p. 141–144.
- Rolf C. Schulz, Rainer Wolf: Copolymerisation zwischen Vinylencarbonat und Isobutylvinyläther. In: Kolloid-Zeitschrift & Zeitschrift für Polymere. 220, 1967, p. 148–151, doi:10.1007/BF02085908.
- M. Le Blanc, K. Andrich: Photobromierung des Toluols. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 20.18‐19, 1914, p. 543–547, doi:10.1002/bbpc.19140201804.
References
- ↑ Jean-Baptiste Dumas: Ueber die Einwirkung des Chlors auf den aus essigsauren Salzen entstehenden Kohlenwasserstoff. In: Annalen der Chemie und Pharmacie. 33, 1840, p. 187–189, doi:10.1002/jlac.18400330205.
- ↑ Jean-Baptiste Dumas: Über das Gesetz der Substitution und die Theorie der Typen, Lieb. Ann., Vol. 33, 1840, p. 259–300.
- ↑ Max Bodenstein: Photochemische Kinetik des Chlorknallgases. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie, 19, 1913, p. 836–856, doi:10.1002/bbpc.19130192104.
- ↑ Franz Rudolf Minz, Reinhard Schliebs: Moderne Verfahren der Großchemie: Chlor und Natronlauge. In: Chemie in unserer Zeit 12. Jahrg. 1978, Nr. 5, ISSN 0009-2851, p. 135–145.
- 1 2 Wilhelm Hirschkind: Chlorination of Saturated Hydrocarbons. In: Industrial & Engineering Chemistry. 41, 1949, p. 2749–2752, doi:10.1021/ie50480a021.
- 1 2 3 United Nations Environment Programme, International Labour Organisation, World Health Organisation, International Programme on Chemical Safety, Environmental Health Criteria 181: CHLORINATED PARAFFINS.
- ↑ Earl T. McBee, Ogden R Pierce: Halogenation. In: Industrial & Engineering Chemistry. 46, 1954, p. 1835–1841, doi:10.1021/ie50537a031.
- ↑ Martin Dameris, Thomas Peter, Ulrich Schmidt, Reinhard Zellner: Das Ozonloch und seine Ursachen, In: Chemie in unserer Zeit, 2007, 41, p. 152–168; doi:10.1002/ciuz.200700418.
- 1 2 Keith U. Ingold, J. Lusztyk, K. D. Raner: The unusual and the unexpected in an old reaction. The photochlorination of alkanes with molecular chlorine in solution. In: Accounts of Chemical Research. 23, 1990, p. 219, doi:10.1021/ar00175a003.
- ↑ Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.): Methoden der organischen Chemie. IV/5a Photochemie. Thieme Verlag, Stuttgart 1975, ISBN 978-3-13-201904-1, p. 91.
- ↑ Max Bodenstein: Sitzung vom 15. Dezember 1930. Berichte der deutschen chemischen Gesellschaft (A and B Series) 64.1 (1931): A1–A4.
- ↑ Glen A. Russell: Solvent Effects in the Reactions of Free Radicals and Atoms. III. Effects of Solvents in the Competitive Photochlorination of Hydrocarbons and Their Derivatives. In: Journal of the American Chemical Society. 80, 1958, p. 4997–5001, doi:10.1021/ja01551a057.
- ↑ D. J. Hurley, R. W. Rosenthal, R. C. Williamson: Effect of Chlorination Conditions on Preparation and Isomer Distribution of Linear Detergent Alkylate. In: Industrial & Engineering Chemistry Product Research and Development. 4, 1965, p. 22, doi:10.1021/i360013a007.
- 1 2 Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.): Methoden der organischen Chemie. IV/5a Photochemie. Thieme Verlag, Stuttgart 1975, ISBN 978-3-13-201904-1, p. 95.
- 1 2 Richard Wegler (Hrsg.): Chemie der Pflanzenschutz und Schädlingsbekämpfungsmittel. Band 1, Springer-Verlag, 1970, ISBN 978-3-642-46212-2, p. 129–132.
- 1 2 Eugen Müller (Hrsg,), E. Forche, W. Hahn: Methoden der organischen Chemie Band V/3: Halogenverbindungen. Fluorverbindungen. Herstellung, Reaktivität und Umwandlung. Chlorverbindungen. Thieme Verlag, 1962, p. 571–573.
- ↑ Directive 2003/53/EC of the European Parliament and of the Council of 18 June 2003 amending for the 26th time Council Directive 76/769/EEC relating to restrictions on the marketing and use of certain dangerous substances and preparations (nonylphenol, nonylphenol ethoxylate and cement)
- ↑ Heinz Strack: Chlorinated paraffins. In: Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim, 1986, VCH Verlagsgesellschaft, Vol. A6, p. 323–330.
- ↑ Heidelore Fiedler (Hrsg.): Chlorinated Paraffins. In: The Handbook of Environmental Chemistry, Springer-Verlag, 2010, ISBN 978-3-642-10760-3, p. 8.
- ↑ Joachim Stauff, H. J. Schumacher: Apparatur zur Untersuchung von Lichtreaktionen der Halogene mit organischen Substanzen: Die Lichtreaktion zwischen Chlor und n‐Heptan. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 48, 1942, p. 271–278, doi:10.1002/bbpc.194200006.
- ↑ Martin Fischer: Industrial Applications of Photochemical Syntheses. In: Angewandte Chemie International Edition in English. 17, 1978, p. 16–26, doi:10.1002/anie.197800161.
- ↑ Roger Adams, A. F. Thal: Benzyl Cyanide. In: Organic Syntheses. 2, 1922, p. 9, doi:10.15227/orgsyn.002.0009.
- ↑ Roger Adams, A. F. Thal: Phenylacetic Acid [α-Toluic acid]. In: Organic Syntheses. 2, 1922, p. 63, doi:10.15227/orgsyn.002.0063.
- ↑ Final Report on the Safety Assessment of Benzaldehyde. In: International Journal of Toxicology. 25, 2006, p. 11–27, doi:10.1080/10915810600716612.
- ↑ Siegfried Hauptmann: Organische Chemie, 2. Auflage, VEB Deutscher Verlag für Grundstoffindustrie, Leipzig, 1985, ISBN 3-342-00280-8, p. 757.
- ↑ Barbara Elvers (Hrsg.): Ullmann’s Encyclopedia of Industrial Chemistry: 7th Edition, Wiley-VCH, 2002, ISBN 978-3-527-30385-4, p. 139.
- ↑ US 2046090A, Reed, Cortes F., "Method of halogenating compounds and product resulting therefrom", published 1933-12-29, issued 1936-06-30
- ↑ US Grant 2174492, Cortes F. Reed, "Preparation of alkane sulphonyl chlorides", issued 1938-09-26, assigned to Charles L Horn
- ↑ Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.): Methoden der organischen Chemie. IV/5a Photochemie. Thieme Verlag, Stuttgart 1975, ISBN 978-3-13-201904-1, p. 165–176.
- ↑ Klaus Schwetlick (2009). Organikum: organisch-chemisches Grundpraktikum (in German) (23rd ed.). Weinheim: Wiley-VCH. p. 206. ISBN 978-3-527-32292-3.
- ↑ Rudolf Bock: Gewinnung von Brom aus Meerwasser. In: Chemie Ingenieur Technik – CIT. 25, 1953, p. 245, doi:10.1002/cite.330250507.
- ↑ Hans-Friedrich Grützmacher, Jürgen Schmiegel: Dithia-diaza[n.2]metacyclophan-ene. In: Chemische Berichte. 122, 1989, p. 1929–1933, doi:10.1002/cber.19891221017.