
In chemistry, an ionic compound is a chemical compound composed of ions held together by electrostatic forces termed ionic bonding. The compound is neutral overall, but consists of positively charged ions called cations and negatively charged ions called anions. These can be simple ions such as the sodium (Na+) and chloride (Cl−) in sodium chloride, or polyatomic species such as the ammonium (NH+
4) and carbonate (CO2−
3) ions in ammonium carbonate. Individual ions within an ionic compound usually have multiple nearest neighbours, so are not considered to be part of molecules, but instead part of a continuous three-dimensional network. Ionic compounds usually form crystalline structures when solid.
Ionic compounds containing basic ions hydroxide (OH−) or oxide (O2−) are classified as bases. Ionic compounds without these ions are also known as salts and can be formed by acid–base reactions. Ionic compounds can also be produced from their constituent ions by evaporation of their solvent, precipitation, freezing, a solid-state reaction, or the electron transfer reaction of reactive metals with reactive non-metals, such as halogen gases.
Ionic compounds typically have high melting and boiling points, and are hard and brittle. As solids they are almost always electrically insulating, but when melted or dissolved they become highly conductive, because the ions are mobilized.
History of discovery
The word ion is the Greek ἰόν, ion, "going", the present participle of ἰέναι, ienai, "to go". This term was introduced by physicist and chemist Michael Faraday in 1834 for the then-unknown species that goes from one electrode to the other through an aqueous medium.[1][2]
.jpg.webp)
In 1913 the crystal structure of sodium chloride was determined by William Henry Bragg and William Lawrence Bragg.[3][4][5] This revealed that there were six equidistant nearest-neighbours for each atom, demonstrating that the constituents were not arranged in molecules or finite aggregates, but instead as a network with long-range crystalline order.[5] Many other inorganic compounds were also found to have similar structural features.[5] These compounds were soon described as being constituted of ions rather than neutral atoms, but proof of this hypothesis was not found until the mid-1920s, when X-ray reflection experiments (which detect the density of electrons), were performed.[5][6]
Principal contributors to the development of a theoretical treatment of ionic crystal structures were Max Born, Fritz Haber, Alfred Landé, Erwin Madelung, Paul Peter Ewald, and Kazimierz Fajans.[7] Born predicted crystal energies based on the assumption of ionic constituents, which showed good correspondence to thermochemical measurements, further supporting the assumption.[5]
Formation

Ionic compounds can be produced from their constituent ions by evaporation, precipitation, or freezing. Reactive metals such as the alkali metals can react directly with the highly electronegative halogen gases to form an ionic product.[8] They can also be synthesized as the product of a high temperature reaction between solids.[9]
If the ionic compound is soluble in a solvent, it can be obtained as a solid compound by evaporating the solvent from this electrolyte solution.[10] As the solvent is evaporated, the ions do not go into the vapor, but stay in the remaining solution, and when they become sufficiently concentrated, nucleation occurs, and they crystallize into an ionic compound. This process occurs widely in nature and is the means of formation of the evaporite minerals.[11] Another method of recovering the compound from solution involves saturating a solution at high temperature and then reducing the solubility by reducing the temperature until the solution is supersaturated and the solid compound nucleates.[10]
Insoluble ionic compounds can be precipitated by mixing two solutions, one with the cation and one with the anion in it. Because all solutions are electrically neutral, the two solutions mixed must also contain counterions of the opposite charges. To ensure that these do not contaminate the precipitated ionic compound, it is important to ensure they do not also precipitate.[12] If the two solutions have hydrogen ions and hydroxide ions as the counterions, they will react with one another in what is called an acid–base reaction or a neutralization reaction to form water.[13] Alternately the counterions can be chosen to ensure that even when combined into a single solution they will remain soluble as spectator ions.[12]
If the solvent is water in either the evaporation or precipitation method of formation, in many cases the ionic crystal formed also includes water of crystallization, so the product is known as a hydrate, and can have very different chemical properties.[14]
Molten salts will solidify on cooling to below their freezing point.[15] This is sometimes used for the solid-state synthesis of complex ionic compounds from solid reactants, which are first melted together.[16] In other cases, the solid reactants do not need to be melted, but instead can react through a solid-state reaction route. In this method, the reactants are repeatedly finely ground into a paste and then heated to a temperature where the ions in neighboring reactants can diffuse together during the time the reactant mixture remains in the oven.[9] Other synthetic routes use a solid precursor with the correct stoichiometric ratio of non-volatile ions, which is heated to drive off other species.[9]
In some reactions between highly reactive metals (usually from Group 1 or Group 2) and highly electronegative halogen gases, or water, the atoms can be ionized by electron transfer,[17] a process thermodynamically understood using the Born–Haber cycle.[18]
Bonding

Ions in ionic compounds are primarily held together by the electrostatic forces between the charge distribution of these bodies, and in particular, the ionic bond resulting from the long-ranged Coulomb attraction between the net negative charge of the anions and net positive charge of the cations.[19] There is also a small additional attractive force from van der Waals interactions which contributes only around 1–2% of the cohesive energy for small ions.[20] When a pair of ions comes close enough for their outer electron shells (most simple ions have closed shells) to overlap, a short-ranged repulsive force occurs,[21] due to the Pauli exclusion principle.[22] The balance between these forces leads to a potential energy well with minimum energy when the nuclei are separated by a specific equilibrium distance.[21]
If the electronic structure of the two interacting bodies is affected by the presence of one another, covalent interactions (non-ionic) also contribute to the overall energy of the compound formed.[23] Ionic compounds are rarely purely ionic, i.e. held together only by electrostatic forces. The bonds between even the most electronegative/electropositive pairs such as those in caesium fluoride exhibit a small degree of covalency.[24][25] Conversely, covalent bonds between unlike atoms often exhibit some charge separation and can be considered to have a partial ionic character.[23] The circumstances under which a compound will have ionic or covalent character can typically be understood using Fajans' rules, which use only charges and the sizes of each ion. According to these rules, compounds with the most ionic character will have large positive ions with a low charge, bonded to a small negative ion with a high charge.[26] More generally HSAB theory can be applied, whereby the compounds with the most ionic character are those consisting of hard acids and hard bases: small, highly charged ions with a high difference in electronegativities between the anion and cation.[27][28] This difference in electronegativities means that the charge separation, and resulting dipole moment, is maintained even when the ions are in contact (the excess electrons on the anions are not transferred or polarized to neutralize the cations).[29]
Structure

Ions typically pack into extremely regular crystalline structures, in an arrangement that minimizes the lattice energy (maximizing attractions and minimizing repulsions). The lattice energy is the summation of the interaction of all sites with all other sites. For unpolarizable spherical ions, only the charges and distances are required to determine the electrostatic interaction energy. For any particular ideal crystal structure, all distances are geometrically related to the smallest internuclear distance. So for each possible crystal structure, the total electrostatic energy can be related to the electrostatic energy of unit charges at the nearest neighboring distance by a multiplicative constant called the Madelung constant[21] that can be efficiently computed using an Ewald sum.[30] When a reasonable form is assumed for the additional repulsive energy, the total lattice energy can be modelled using the Born–Landé equation,[31] the Born–Mayer equation, or in the absence of structural information, the Kapustinskii equation.[32]
Using an even simpler approximation of the ions as impenetrable hard spheres, the arrangement of anions in these systems are often related to close-packed arrangements of spheres, with the cations occupying tetrahedral or octahedral interstices.[33][34] Depending on the stoichiometry of the ionic compound, and the coordination (principally determined by the radius ratio) of cations and anions, a variety of structures are commonly observed,[35] and theoretically rationalized by Pauling's rules.[36]
| Stoichiometry | Cation:anion coordination |
Interstitial sites | Cubic close packing of anions | Hexagonal close packing of anions | |||
|---|---|---|---|---|---|---|---|
| Occupancy | Critical radius ratio |
Name | Madelung constant | Name | Madelung constant | ||
| MX | 6:6 | all octahedral | 0.4142[33] | sodium chloride | 1.747565[37] | nickeline | <1.73[lower-alpha 1][38] |
| 4:4 | alternate tetrahedral | 0.2247[39] | zinc blende | 1.6381[37] | wurtzite | 1.641[5] | |
| MX2 | 8:4 | all tetrahedral | 0.2247 | fluorite | 5.03878[40] | ||
| 6:3 | half octahedral (alternate layers fully occupied) | 0.4142 | cadmium chloride | 5.61[41] | cadmium iodide | 4.71[40] | |
| MX3 | 6:2 | one-third octahedral | 0.4142 | rhodium(III) bromide[lower-alpha 2][42][43] | 6.67[44][lower-alpha 3] | bismuth iodide | 8.26[44][lower-alpha 4] |
| M2X3 | 6:4 | two-thirds octahedral | 0.4142 | corundum | 25.0312[40] | ||
| ABO3 | two-thirds octahedral | 0.4142 | ilmenite | Depends on charges and structure [lower-alpha 5] | |||
| AB2O4 | one-eighth tetrahedral and one-half octahedral | rA/rO = 0.2247, rB/rO = 0.4142[lower-alpha 6] | spinel, inverse spinel | Depends on cation site distributions[47][48][49] | olivine | Depends on cation site distributions[50] | |
In some cases, the anions take on a simple cubic packing and the resulting common structures observed are:
| Stoichiometry | Cation:anion coordination |
Interstitial sites occupied | Example structure | ||
|---|---|---|---|---|---|
| Name | Critical radius ratio |
Madelung constant | |||
| MX | 8:8 | entirely filled | cesium chloride | 0.7321[51] | 1.762675[37] |
| MX2 | 8:4 | half filled | calcium fluoride | ||
| M2X | 4:8 | half filled | lithium oxide | ||
Some ionic liquids, particularly with mixtures of anions or cations, can be cooled rapidly enough that there is not enough time for crystal nucleation to occur, so an ionic glass is formed (with no long-range order).[52]
Defects

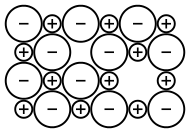
Within an ionic crystal, there will usually be some point defects, but to maintain electroneutrality, these defects come in pairs.[53] Frenkel defects consist of a cation vacancy paired with a cation interstitial and can be generated anywhere in the bulk of the crystal,[53] occurring most commonly in compounds with a low coordination number and cations that are much smaller than the anions.[54] Schottky defects consist of one vacancy of each type, and are generated at the surfaces of a crystal,[53] occurring most commonly in compounds with a high coordination number and when the anions and cations are of similar size.[54] If the cations have multiple possible oxidation states, then it is possible for cation vacancies to compensate for electron deficiencies on cation sites with higher oxidation numbers, resulting in a non-stoichiometric compound.[53] Another non-stoichiometric possibility is the formation of an F-center, a free electron occupying an anion vacancy.[55] When the compound has three or more ionic components, even more defect types are possible.[53] All of these point defects can be generated via thermal vibrations and have an equilibrium concentration. Because they are energetically costly but entropically beneficial, they occur in greater concentration at higher temperatures. Once generated, these pairs of defects can diffuse mostly independently of one another, by hopping between lattice sites. This defect mobility is the source of most transport phenomena within an ionic crystal, including diffusion and solid state ionic conductivity.[53] When vacancies collide with interstitials (Frenkel), they can recombine and annihilate one another. Similarly, vacancies are removed when they reach the surface of the crystal (Schottky). Defects in the crystal structure generally expand the lattice parameters, reducing the overall density of the crystal.[53] Defects also result in ions in distinctly different local environments, which causes them to experience a different crystal-field symmetry, especially in the case of different cations exchanging lattice sites.[53] This results in a different splitting of d-electron orbitals, so that the optical absorption (and hence colour) can change with defect concentration.[53]
Properties
Acidity/basicity
Ionic compounds containing hydrogen ions (H+) are classified as acids, and those containing electropositive cations[56] and basic anions ions hydroxide (OH−) or oxide (O2−) are classified as bases. Other ionic compounds are known as salts and can be formed by acid–base reactions.[57] If the compound is the result of a reaction between a strong acid and a weak base, the result is an acidic salt. If it is the result of a reaction between a strong base and a weak acid, the result is a basic salt. If it is the result of a reaction between a strong acid and a strong base, the result is a neutral salt. Weak acids reacted with weak bases can produce ionic compounds with both the conjugate base ion and conjugate acid ion, such as ammonium acetate.
Some ions are classed as amphoteric, being able to react with either an acid or a base.[58] This is also true of some compounds with ionic character, typically oxides or hydroxides of less-electropositive metals (so the compound also has significant covalent character), such as zinc oxide, aluminium hydroxide, aluminium oxide and lead(II) oxide.[59]
Melting and boiling points
Electrostatic forces between particles are strongest when the charges are high, and the distance between the nuclei of the ions is small. In such cases, the compounds generally have very high melting and boiling points and a low vapour pressure.[60] Trends in melting points can be even better explained when the structure and ionic size ratio is taken into account.[61] Above their melting point, ionic solids melt and become molten salts (although some ionic compounds such as aluminium chloride and iron(III) chloride show molecule-like structures in the liquid phase).[62] Inorganic compounds with simple ions typically have small ions, and thus have high melting points, so are solids at room temperature. Some substances with larger ions, however, have a melting point below or near room temperature (often defined as up to 100 °C), and are termed ionic liquids.[63] Ions in ionic liquids often have uneven charge distributions, or bulky substituents like hydrocarbon chains, which also play a role in determining the strength of the interactions and propensity to melt.[64]
Even when the local structure and bonding of an ionic solid is disrupted sufficiently to melt it, there are still strong long-range electrostatic forces of attraction holding the liquid together and preventing ions boiling to form a gas phase.[65] This means that even room temperature ionic liquids have low vapour pressures, and require substantially higher temperatures to boil.[65] Boiling points exhibit similar trends to melting points in terms of the size of ions and strength of other interactions.[65] When vapourized, the ions are still not freed of one another. For example, in the vapour phase sodium chloride exists as diatomic "molecules".[66]
Brittleness
Most ionic compounds are very brittle. Once they reach the limit of their strength, they cannot deform malleably, because the strict alignment of positive and negative ions must be maintained. Instead the material undergoes fracture via cleavage.[67] As the temperature is elevated (usually close to the melting point) a ductile–brittle transition occurs, and plastic flow becomes possible by the motion of dislocations.[67][68]
Compressibility
The compressibility of an ionic compound is strongly determined by its structure, and in particular the coordination number. For example, halides with the caesium chloride structure (coordination number 8) are less compressible than those with the sodium chloride structure (coordination number 6), and less again than those with a coordination number of 4.[69]
Solubility
When ionic compounds dissolve, the individual ions dissociate and are solvated by the solvent and dispersed throughout the resulting solution.[70] Because the ions are released into solution when dissolved, and can conduct charge, soluble ionic compounds are the most common class of strong electrolytes, and their solutions have a high electrical conductivity.[71]

The solubility is highest in polar solvents (such as water) or ionic liquids, but tends to be low in nonpolar solvents (such as petrol/gasoline).[72] This is principally because the resulting ion–dipole interactions are significantly stronger than ion-induced dipole interactions, so the heat of solution is higher. When the oppositely charged ions in the solid ionic lattice are surrounded by the opposite pole of a polar molecule, the solid ions are pulled out of the lattice and into the liquid. If the solvation energy exceeds the lattice energy, the negative net enthalpy change of solution provides a thermodynamic drive to remove ions from their positions in the crystal and dissolve in the liquid. In addition, the entropy change of solution is usually positive for most solid solutes like ionic compounds, which means that their solubility increases when the temperature increases.[73] There are some unusual ionic compounds such as cerium(III) sulfate, where this entropy change is negative, due to extra order induced in the water upon solution, and the solubility decreases with temperature.[73]
Electrical conductivity
Although ionic compounds contain charged atoms or clusters, these materials do not typically conduct electricity to any significant extent when the substance is solid. In order to conduct, the charged particles must be mobile rather than stationary in a crystal lattice. This is achieved to some degree at high temperatures when the defect concentration increases the ionic mobility and solid state ionic conductivity is observed. When the ionic compounds are dissolved in a liquid or are melted into a liquid, they can conduct electricity because the ions become completely mobile.[74] This conductivity gain upon dissolving or melting is sometimes used as a defining characteristic of ionic compounds.[75]
In some unusual ionic compounds: fast ion conductors, and ionic glasses,[52] one or more of the ionic components has a significant mobility, allowing conductivity even while the material as a whole remains solid.[76] This is often highly temperature dependent, and may be the result of either a phase change or a high defect concentration.[76] These materials are used in all solid-state supercapacitors, batteries, and fuel cells, and in various kinds of chemical sensors.[77][78]
Colour
_chloride.jpg.webp)
-chloride-hexahydrate-sample.jpg.webp)
CoCl2·6H2O
The colour of an ionic compound is often different from the colour of an aqueous solution containing the constituent ions,[79] or the hydrated form of the same compound.[14]
The anions in compounds with bonds with the most ionic character tend to be colorless (with an absorption band in the ultraviolet part of the spectrum).[80] In compounds with less ionic character, their color deepens through yellow, orange, red, and black (as the absorption band shifts to longer wavelengths into the visible spectrum). [80]
The absorption band of simple cations shifts toward a shorter wavelength when they are involved in more covalent interactions.[80] This occurs during hydration of metal ions, so colorless anhydrous ionic compounds with an anion absorbing in the infrared can become colorful in solution.[80]
Uses
Ionic compounds have long had a wide variety of uses and applications. Many minerals are ionic.[81] Humans have processed common salt (sodium chloride) for over 8000 years, using it first as a food seasoning and preservative, and now also in manufacturing, agriculture, water conditioning, for de-icing roads, and many other uses.[82] Many ionic compounds are so widely used in society that they go by common names unrelated to their chemical identity. Examples of this include borax, calomel, milk of magnesia, muriatic acid, oil of vitriol, saltpeter, and slaked lime.[83]
Soluble ionic compounds like salt can easily be dissolved to provide electrolyte solutions. This is a simple way to control the concentration and ionic strength. The concentration of solutes affects many colligative properties, including increasing the osmotic pressure, and causing freezing-point depression and boiling-point elevation.[84] Because the solutes are charged ions they also increase the electrical conductivity of the solution.[85] The increased ionic strength reduces the thickness of the electrical double layer around colloidal particles, and therefore the stability of emulsions and suspensions.[86]
The chemical identity of the ions added is also important in many uses. For example, fluoride containing compounds are dissolved to supply fluoride ions for water fluoridation.[87]
Solid ionic compounds have long been used as paint pigments, and are resistant to organic solvents, but are sensitive to acidity or basicity.[88] Since 1801 pyrotechnicians have described and widely used metal-containing ionic compounds as sources of colour in fireworks.[89] Under intense heat, the electrons in the metal ions or small molecules can be excited.[90] These electrons later return to lower energy states, and release light with a colour spectrum characteristic of the species present.[91][92]
In chemistry, ionic compounds are often used as precursors for high-temperature solid-state synthesis.[93]
Many metals are geologically most abundant as ionic compounds within ores.[94] To obtain the elemental materials, these ores are processed by smelting or electrolysis, in which redox reactions occur (often with a reducing agent such as carbon) such that the metal ions gain electrons to become neutral atoms.[95][96]
Nomenclature
According to the nomenclature recommended by IUPAC, ionic compounds are named according to their composition, not their structure.[97] In the most simple case of a binary ionic compound with no possible ambiguity about the charges and thus the stoichiometry, the common name is written using two words.[98] The name of the cation (the unmodified element name for monatomic cations) comes first, followed by the name of the anion.[99][100] For example, MgCl2 is named magnesium chloride, and Na2SO4 is named sodium sulfate (SO2−
4, sulfate, is an example of a polyatomic ion). To obtain the empirical formula from these names, the stoichiometry can be deduced from the charges on the ions, and the requirement of overall charge neutrality.[101]
If there are multiple different cations and/or anions, multiplicative prefixes (di-, tri-, tetra-, ...) are often required to indicate the relative compositions,[102] and cations then anions are listed in alphabetical order.[103] For example, KMgCl3 is named magnesium potassium trichloride to distinguish it from K2MgCl4, magnesium dipotassium tetrachloride[104] (note that in both the empirical formula and the written name, the cations appear in alphabetical order, but the order varies between them because the symbol for potassium is K).[105] When one of the ions already has a multiplicative prefix within its name, the alternate multiplicative prefixes (bis-, tris-, tetrakis-, ...) are used.[106] For example, Ba(BrF4)2 is named barium bis(tetrafluoridobromate).[107]
Compounds containing one or more elements which can exist in a variety of charge/oxidation states will have a stoichiometry that depends on which oxidation states are present, to ensure overall neutrality. This can be indicated in the name by specifying either the oxidation state of the elements present, or the charge on the ions.[107] Because of the risk of ambiguity in allocating oxidation states, IUPAC prefers direct indication of the ionic charge numbers.[107] These are written as an arabic integer followed by the sign (... , 2−, 1−, 1+, 2+, ...) in parentheses directly after the name of the cation (without a space separating them).[107] For example, FeSO4 is named iron(2+) sulfate (with the 2+ charge on the Fe2+ ions balancing the 2− charge on the sulfate ion), whereas Fe2(SO4)3 is named iron(3+) sulfate (because the two iron ions in each formula unit each have a charge of 3+, to balance the 2− on each of the three sulfate ions).[107] Stock nomenclature, still in common use, writes the oxidation number in Roman numerals (... , −II, −I, 0, I, II, ...). So the examples given above would be named iron(II) sulfate and iron(III) sulfate respectively.[108] For simple ions the ionic charge and the oxidation number are identical, but for polyatomic ions they often differ. For example, the uranyl(2+) ion, UO2+
2, has uranium in an oxidation state of +6, so would be called a dioxouranium(VI) ion in Stock nomenclature.[109] An even older naming system for metal cations, also still widely used, appended the suffixes -ous and -ic to the Latin root of the name, to give special names for the low and high oxidation states.[110] For example, this scheme uses "ferrous" and "ferric", for iron(II) and iron(III) respectively,[110] so the examples given above were classically named ferrous sulfate and ferric sulfate.
See also
Notes
- ↑ This structure type has a variable lattice parameter c/a ratio, and the exact Madelung constant depends on this.
- ↑ This structure has been referred to in references as yttrium(III) chloride and chromium(III) chloride, but both are now known as the RhBr3 structure type.
- ↑ The reference lists this structure as MoCl3, which is now known as the RhBr3 structure.
- ↑ The reference lists this structure as FeCl3, which is now known as the BiI3 structure type.
- ↑ This structure type can accommodate any charges on A and B that add up to six. When both are three the charge structure is equivalent to that of corrundum.[45] The structure also has a variable lattice parameter c/a ratio, and the exact Madelung constant depends on this.
- ↑ However, in some cases such as MgAl2O4 the larger cation occupies the smaller tetrahedral site.[46]
References
- ↑ Michael Faraday (1791–1867). UK: BBC. Archived from the original on 2016-08-25.
- ↑ "Online etymology dictionary". Archived from the original on 2011-05-14. Retrieved 2011-01-07.
- ↑ Bragg, W. H.; Bragg, W. L. (1 July 1913). "The Reflection of X-rays by Crystals". Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences. 88 (605): 428–438. Bibcode:1913RSPSA..88..428B. doi:10.1098/rspa.1913.0040. S2CID 13112732.
- ↑ Bragg, W. H. (22 September 1913). "The Reflection of X-rays by Crystals. (II.)". Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences. 89 (610): 246–248. Bibcode:1913RSPSA..89..246B. doi:10.1098/rspa.1913.0082.
- 1 2 3 4 5 6 Sherman, Jack (August 1932). "Crystal Energies of Ionic Compounds and Thermochemical Applications". Chemical Reviews. 11 (1): 93–170. doi:10.1021/cr60038a002.
- ↑ James, R. W.; Brindley, G. W. (1 November 1928). "A Quantitative Study of the Reflexion of X-Rays by Sylvine". Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences. 121 (787): 155–171. Bibcode:1928RSPSA.121..155J. doi:10.1098/rspa.1928.0188.
- ↑ Pauling 1960, p. 505.
- ↑ Zumdahl 1989, p. 312.
- 1 2 3 Wold & Dwight 1993, p. 71.
- 1 2 Wold & Dwight 1993, p. 82.
- ↑ Wenk, Hans-Rudolf; Bulakh, Andrei (2003). Minerals: their constitution and origin (Reprinted with corrections. ed.). New York: Cambridge University Press. p. 351. ISBN 978-0-521-52958-7. Archived from the original on 2017-12-03.
- 1 2 Zumdahl 1989, p. 133–140.
- ↑ Zumdahl 1989, p. 144–145.
- 1 2 Brown 2009, p. 417.
- ↑ Wold & Dwight 1993, p. 79.
- ↑ Wold & Dwight 1993, pp. 79–81.
- ↑ Zumdahl 1989, p. 312–313.
- ↑ Barrow 1988, p. 161–162.
- ↑ Pauling 1960, p. 6.
- ↑ Kittel 2005, p. 61.
- 1 2 3 Pauling 1960, p. 507.
- ↑ Ashcroft & Mermin 1977, p. 379.
- 1 2 Pauling 1960, p. 65.
- ↑ Hannay, N. Bruce; Smyth, Charles P. (February 1946). "The Dipole Moment of Hydrogen Fluoride and the Ionic Character of Bonds". Journal of the American Chemical Society. 68 (2): 171–173. doi:10.1021/ja01206a003.
- ↑ Pauling, Linus (1948). "The modern theory of valency". Journal of the Chemical Society (Resumed). 17: 1461–1467. doi:10.1039/JR9480001461. PMID 18893624. Archived from the original on 2021-12-07. Retrieved 2021-12-01.
- ↑ Lalena, John. N.; Cleary, David. A. (2010). Principles of inorganic materials design (2nd ed.). Hoboken, N.J: John Wiley. ISBN 978-0-470-56753-1.
- ↑ Pearson, Ralph G. (November 1963). "Hard and Soft Acids and Bases". Journal of the American Chemical Society. 85 (22): 3533–3539. doi:10.1021/ja00905a001.
- ↑ Pearson, Ralph G. (October 1968). "Hard and soft acids and bases, HSAB, part II: Underlying theories". Journal of Chemical Education. 45 (10): 643. Bibcode:1968JChEd..45..643P. doi:10.1021/ed045p643.
- ↑ Barrow 1988, p. 676.
- ↑ Kittel 2005, p. 64.
- ↑ Pauling 1960, p. 509.
- ↑ Carter, Robert (2016). "Lattice Energy" (PDF). CH370 Lecture Material. Archived (PDF) from the original on 2015-05-13. Retrieved 2016-01-19.
- 1 2 Ashcroft & Mermin 1977, p. 383.
- ↑ Zumdahl 1989, p. 444–445.
- 1 2 Moore, Lesley E. Smart; Elaine A. (2005). Solid state chemistry: an introduction (3. ed.). Boca Raton, Fla. [u.a.]: Taylor & Francis, CRC. p. 44. ISBN 978-0-7487-7516-3.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ↑ Ashcroft & Mermin 1977, pp. 382–387.
- 1 2 3 Kittel 2005, p. 65.
- ↑ Zemann, J. (1 January 1958). "Berechnung von Madelung'schen Zahlen für den NiAs-Typ". Acta Crystallographica. 11 (1): 55–56. doi:10.1107/S0365110X5800013X.
- ↑ Ashcroft & Mermin 1977, p. 386.
- 1 2 3 Dienes, Richard J. Borg, G.J. (1992). The physical chemistry of solids. Boston: Academic Press. p. 123. ISBN 978-0-12-118420-9.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ↑ Brackett, Thomas E.; Brackett, Elizabeth B. (1965). "The Lattice Energies of the Alkaline Earth Halides". Journal of Physical Chemistry. 69 (10): 3611–3614. doi:10.1021/j100894a062.
- ↑ "YCl3 – Yttrium trichloride". ChemTube3D. University of Liverpool. 2008. Archived from the original on 27 January 2016. Retrieved 19 January 2016.
- 1 2 Ellis, Arthur B. []; et al. (1995). Teaching general chemistry: a materials science companion (3. print ed.). Washington: American Chemical Society. p. 121. ISBN 978-0-8412-2725-5.
- 1 2 Hoppe, R. (January 1966). "Madelung Constants". Angewandte Chemie International Edition in English. 5 (1): 95–106. doi:10.1002/anie.196600951.
- ↑ Bhagi, Ajay; Raj, Gurdeep (2010). Krishna's IAS Chemistry. Meerut: Krishna Prakashan Media. p. 171. ISBN 978-81-87224-70-9.
- ↑ Wenk & Bulakh 2004, p. 778.
- ↑ Verwey, E. J. W. (1947). "Physical Properties and Cation Arrangement of Oxides with Spinel Structures I. Cation Arrangement in Spinels". Journal of Chemical Physics. 15 (4): 174–180. Bibcode:1947JChPh..15..174V. doi:10.1063/1.1746464.
- ↑ Verwey, E. J. W.; de Boer, F.; van Santen, J. H. (1948). "Cation Arrangement in Spinels". The Journal of Chemical Physics. 16 (12): 1091. Bibcode:1948JChPh..16.1091V. doi:10.1063/1.1746736.
- ↑ Thompson, P.; Grimes, N. W. (27 September 2006). "Madelung calculations for the spinel structure". Philosophical Magazine. Vol. 36, no. 3. pp. 501–505. Bibcode:1977PMag...36..501T. doi:10.1080/14786437708239734.
- ↑ Alberti, A.; Vezzalini, G. (1978). "Madelung energies and cation distributions in olivine-type structures". Zeitschrift für Kristallographie – Crystalline Materials. 147 (1–4): 167–176. Bibcode:1978ZK....147..167A. doi:10.1524/zkri.1978.147.14.167. hdl:11380/738457. S2CID 101158673.
- ↑ Ashcroft & Mermin 1977, p. 384.
- 1 2 Souquet, J (October 1981). "Electrochemical properties of ionically conductive glasses". Solid State Ionics. 5: 77–82. doi:10.1016/0167-2738(81)90198-3.
- 1 2 3 4 5 6 7 8 9 Schmalzried, Hermann (1965). "Point defects in ternary ionic crystals". Progress in Solid State Chemistry. 2: 265–303. doi:10.1016/0079-6786(65)90009-9.
- 1 2 Prakash, Satya (1945). Advanced inorganic chemistry. New Delhi: S. Chand & Company Ltd. p. 554. ISBN 978-81-219-0263-2.
- ↑ Kittel 2005, p. 376.
- ↑ "Periodic Trends and Oxides". Archived from the original on 2015-12-29. Retrieved 2015-11-10.
- ↑ Whitten, Kenneth W.; Galley, Kenneth D.; Davis, Raymond E. (1992). General Chemistry (4th ed.). Saunders. p. 128. ISBN 978-0-03-072373-5.
- ↑ Davidson, David (November 1955). "Amphoteric molecules, ions and salts". Journal of Chemical Education. 32 (11): 550. Bibcode:1955JChEd..32..550D. doi:10.1021/ed032p550.
- ↑ Weller, Mark; Overton, Tina; Rourke, Jonathan; Armstrong, Fraser (2014). Inorganic chemistry (Sixth ed.). Oxford: Oxford University Press. pp. 129–130. ISBN 978-0-19-964182-6.
- ↑ McQuarrie & Rock 1991, p. 503.
- ↑ Pauling, Linus (1928-04-01). "The Influence of Relative Ionic Sizes on the Properties of Ionic Compounds". Journal of the American Chemical Society. 50 (4): 1036–1045. doi:10.1021/ja01391a014. ISSN 0002-7863.
- ↑ Tosi, M. P. (2002). Gaune-Escard, Marcelle (ed.). Molten Salts: From Fundamentals to Applications. Dordrecht: Springer Netherlands. p. 1. ISBN 978-94-010-0458-9. Archived from the original on 2017-12-03.
- ↑ Freemantle 2009, p. 1.
- ↑ Freemantle 2009, pp. 3–4.
- 1 2 3 Rebelo, Luis P. N.; Canongia Lopes, José N.; Esperança, José M. S. S.; Filipe, Eduardo (2005-04-01). "On the Critical Temperature, Normal Boiling Point, and Vapor Pressure of Ionic Liquids". The Journal of Physical Chemistry B. 109 (13): 6040–6043. doi:10.1021/jp050430h. ISSN 1520-6106. PMID 16851662.
- ↑ Porterfield, William W. (2013). Inorganic Chemistry a Unified Approach (2nd ed.). New York: Elsevier Science. pp. 63–67. ISBN 978-0-323-13894-9. Archived from the original on 2017-12-03.
- 1 2 Johnston, T. L.; Stokes, R. J.; Li, C. H. (December 1959). "The ductile–brittle transition in ionic solids". Philosophical Magazine. Vol. 4, no. 48. pp. 1316–1324. Bibcode:1959PMag....4.1316J. doi:10.1080/14786435908233367.
- ↑ Kelly, A.; Tyson, W. R.; Cottrell, A. H. (1967-03-01). "Ductile and brittle crystals". Philosophical Magazine. Vol. 15, no. 135. pp. 567–586. Bibcode:1967PMag...15..567K. doi:10.1080/14786436708220903. ISSN 0031-8086.
- ↑ Stillwell, Charles W. (January 1937). "Crystal chemistry. V. The properties of binary compounds". Journal of Chemical Education. 14 (1): 34. Bibcode:1937JChEd..14...34S. doi:10.1021/ed014p34.
- ↑ Brown 2009, pp. 89–91.
- ↑ Brown 2009, pp. 91–92.
- ↑ Brown 2009, pp. 413–415.
- 1 2 Brown 2009, p. 422.
- ↑ "Electrical Conductivity of Ionic Compound". 2011-05-22. Archived from the original on 21 May 2014. Retrieved 2 December 2012.
- ↑ Zumdahl 1989, p. 341.
- 1 2 Gao, Wei; Sammes, Nigel M (1999). An Introduction to Electronic and Ionic Materials. World Scientific. p. 261. ISBN 978-981-02-3473-7. Archived from the original on 2017-12-03.
- ↑ West, Anthony R. (1991). "Solid electrolytes and mixed ionic?electronic conductors: an applications overview". Journal of Materials Chemistry. 1 (2): 157. doi:10.1039/JM9910100157.
- ↑ Boivin, J. C.; Mairesse, G. (October 1998). "Recent Material Developments in Fast Oxide Ion Conductors". Chemistry of Materials. 10 (10): 2870–2888. doi:10.1021/cm980236q.
- ↑ Pauling 1960, p. 105.
- 1 2 3 4 Pauling 1960, p. 107.
- ↑ Wenk & Bulakh 2004, p. 774.
- ↑ Kurlansky, Mark (2003). Salt: a world history (1st ed.). London: Vintage. ISBN 978-0-09-928199-3.
- ↑ Lower, Simon (2014). "Naming Chemical Substances". Chem1 General Chemistry Virtual Textbook. Archived from the original on 16 January 2016. Retrieved 14 January 2016.
- ↑ Atkins & de Paula 2006, pp. 150–157.
- ↑ Atkins & de Paula 2006, pp. 761–770.
- ↑ Atkins & de Paula 2006, pp. 163–169.
- ↑ Reeves TG (1986). "Water fluoridation: a manual for engineers and technicians" (PDF). Centers for Disease Control. Archived from the original (PDF) on 2017-02-08. Retrieved 2016-01-18.
- ↑ Satake, M; Mido, Y (1995). Chemistry of Colour. Discovery Publishing House. p. 230. ISBN 978-81-7141-276-1. Archived from the original on 2017-12-03.
- ↑ Russell 2009, p. 14.
- ↑ Russell 2009, p. 82.
- ↑ Russell 2009, pp. 108–117.
- ↑ Russell 2009, pp. 129–133.
- ↑ Xu, Ruren; Pang, Wenqin; Huo, Qisheng (2011). Modern inorganic synthetic chemistry. Amsterdam: Elsevier. p. 22. ISBN 978-0-444-53599-3.
- ↑ Zumdahl & Zumdahl 2015, pp. 822.
- ↑ Zumdahl & Zumdahl 2015, pp. 823.
- ↑ Gupta, Chiranjib Kumar (2003). Chemical metallurgy principles and practice. Weinheim: Wiley-VCH. pp. 359–365. ISBN 978-3-527-60525-5.
- ↑ IUPAC 2005, p. 68.
- ↑ IUPAC 2005, p. 70.
- ↑ IUPAC 2005, p. 69.
- ↑ Kotz, John C.; Treichel, Paul M; Weaver, Gabriela C. (2006). Chemistry and Chemical Reactivity (Sixth ed.). Belmont, CA: Thomson Brooks/Cole. p. 111. ISBN 978-0-534-99766-3.
- ↑ Brown 2009, pp. 36–37.
- ↑ IUPAC 2005, pp. 75–76.
- ↑ IUPAC 2005, p. 75.
- ↑ Gibbons, Cyril S.; Reinsborough, Vincent C.; Whitla, W. Alexander (January 1975). "Crystal Structures of K2MgCl4 and Cs2MgCl4". Canadian Journal of Chemistry. 53 (1): 114–118. doi:10.1139/v75-015.
- ↑ IUPAC 2005, p. 76.
- ↑ IUPAC 2005, pp. 76–77.
- 1 2 3 4 5 IUPAC 2005, p. 77.
- ↑ IUPAC 2005, pp. 77–78.
- ↑ Fernelius, W. Conard (November 1982). "Numbers in chemical names". Journal of Chemical Education. 59 (11): 964. Bibcode:1982JChEd..59..964F. doi:10.1021/ed059p964.
- 1 2 Brown 2009, p. 38.
Bibliography
- Ashcroft, Neil W.; Mermin, N. David (1977). Solid state physics (27th repr. ed.). New York: Holt, Rinehart and Winston. ISBN 978-0-03-083993-1.
- Atkins, Peter; de Paula, Julio (2006). Atkins' physical chemistry (8th ed.). Oxford: Oxford University Press. ISBN 978-0-19-870072-2.
- Barrow, Gordon M. (1988). Physical chemistry (5th ed.). New York: McGraw-Hill. ISBN 978-0-07-003905-6.
- Brown, Theodore L.; LeMay, H. Eugene Jr; Bursten, Bruce E.; Lanford, Steven; Sagatys, Dalius; Duffy, Neil (2009). Chemistry: the central science: a broad perspective (2nd ed.). Frenchs Forest, N.S.W.: Pearson Australia. ISBN 978-1-4425-1147-7.
- Freemantle, Michael (2009). An introduction to ionic liquids. Cambridge: Royal Society of Chemistry. ISBN 978-1-84755-161-0.
- International Union of Pure and Applied Chemistry, Division of Chemical Nomenclature (2005). Neil G. Connelly (ed.). Nomenclature of inorganic chemistry: IUPAC recommendations 2005 (New ed.). Cambridge: RSC Publ. ISBN 978-0-85404-438-2. Archived from the original on 2016-02-03. Retrieved 2023-02-05.
- Kittel, Charles (2005). Introduction to Solid State Physics (8th ed.). Hoboken, NJ: John Wiley & Sons. ISBN 978-0-471-41526-8.
- McQuarrie, Donald A.; Rock, Peter A. (1991). General chemistry (3rd ed.). New York: W.H. Freeman and Co. ISBN 978-0-7167-2169-7.
- Pauling, Linus (1960). The nature of the chemical bond and the structure of molecules and crystals: an introduction to modern structural chemistry (3rd ed.). Ithaca, N.Y.: Cornell University Press. ISBN 978-0-8014-0333-0.
- Russell, Michael S. (2009). The chemistry of fireworks (2nd ed.). Cambridge, UK: RSC Pub. ISBN 978-0-85404-127-5.
- Wenk, Hans-Rudolph; Bulakh, Andrei (2004). Minerals: Their Constitution and Origin (1st ed.). New York: Cambridge University Press. ISBN 978-1-107-39390-5.
- Wold, Aaron; Dwight, Kirby (1993). Solid State Chemistry Synthesis, Structure, and Properties of Selected Oxides and Sulfides. Dordrecht: Springer Netherlands. ISBN 978-94-011-1476-9.
- Zumdahl, Steven S. (1989). Chemistry (2nd ed.). Lexington, Mass.: D.C. Heath. ISBN 978-0-669-16708-5.
- Zumdahl, Steven; Zumdahl, Susan (2015). Chemistry: An Atoms First Approach. Cengage Learning. ISBN 978-1-305-68804-9.