| Glycerol kinase deficiency | |
|---|---|
| Other names | GKD |
Glycerol kinase deficiency (GKD) is an X-linked recessive enzyme defect that is heterozygous in nature. Three clinically distinct forms of this deficiency have been proposed, namely infantile, juvenile, and adult. National Institutes of Health and its Office of Rare Diseases Research branch classifies GKD as a rare disease, known to affect fewer than 200,000 individuals in the United States. The responsible gene lies in a region containing genes in which deletions can cause Duchenne muscular dystrophy and adrenal hypoplasia congenita. Combinations of these three genetic defects including GKD are addressed medically as Complex GKD.[1]
Signs and symptoms
Glycerol Kinase Deficiency causes the condition known as hyperglycerolemia,[2] an accumulation of glycerol in the blood and urine. This excess of glycerol in bodily fluids can lead to many more potentially dangerous symptoms. Common symptoms include vomiting and lethargy.[3] These tend to be the only symptoms, if any, present in adult GKD which has been found to present with fewer symptoms than infant or juvenile GKD.[4] When GKD is accompanied by Duchenne muscular dystrophy and Adrenal Hypoplasia Congenita, also caused by mutations on the Xp21 chromosome,[5] the symptoms can become much more severe. Symptoms visible at or shortly after birth include:
- cryptorchidism
- strabismus
- seizures[3]
Some other symptoms that become more noticeable with time would be:
- metabolic acidosis
- hypoglycemia
- adrenal cortex insufficiency[6]
- learning disabilities
- osteoporosis
- myopathy
Many of the physically visible symptoms, such as cryptorchidism, strabismus, learning disabilities,[5] and myopathy, tend to have an added psychological effect on the subject due to the fact that they can set him or her apart from those without GKD. Cryptorchidism, the failure of one or both of the testes to descend to the scrotum, has been known to lead to sexual identity confusion amongst young boys because it is such a major physiological anomaly.[7] Strabismus is the misalignment of one's eyes. Typically, one is focused but the other is “lazy” and is directed inward or out ward (up and down is less common but does occur).
Causes
Glycerol kinase deficiency has two main causes.
- The first cause is isolated enzyme deficiency. The enzyme glycerol kinase is encoded by the X-chromosome in humans.[8] It acts as a catalyst in the phosphorylation of glycerol to glycerol-3-phosphate which plays a key role in formation of triacylglycerol (TAG) and fat storage. There is no strict genotype–phenotype correlation in isolated GKD; it can be either symptomatic or asymptomatic.[9] Symptomatic means that GKD shows symptoms when it persists in the body and asymptomatic means that the no symptoms appear in the body. In this deficiency the genotype is not associated with the phenotype. The presence of certain mutations in genes has no relation with the phenotype i.e. any resulting physical traits or abnormality.[10]
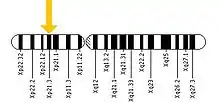 Location of the Glycerol Kinase gene on the X chromosome
Location of the Glycerol Kinase gene on the X chromosome - The second cause is a deletion or mutation of a single gene. GKD is described by Mendelian inheritance and is an X-linked recessive trait due to which it occurs mainly in males and occasionally in females.[11] GKD results when the glycerol kinase gene present on the locus Xp21 of the X chromosome is either deleted or mutated. Females have two X chromosomes and males have one X and one Y chromosome. The expression of recessive genes on the X chromosome is different in males and females. This is due to the fact that genes present on the Y chromosome do not pair up with genes on the X chromosome in males. In females the disorder is expressed only when there are two copies of the affected gene present on each X chromosome but since the glycerol kinase gene is present only on one X chromosome the disorder is not expressed in women. Women have a second good copy that can compensate for the defect on the first copy. On the other hand, males only need a single copy of the recessive gene for the disorder to be expressed. They do not have a second copy that can protect against any defect on the first copy.[12]
Effect on glycolysis
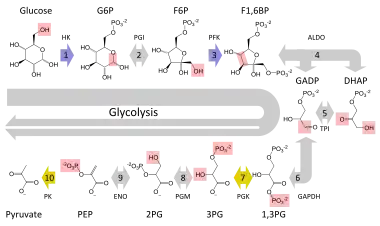
In order to understand how this condition affects a person you must first have a basic understanding of the process called glycolysis. This fundamental metabolic pathway is found in all known organisms. The process provides energy for our cells to carry out their daily functions. The overall reaction involves a cell taking in the sugar glucose and converting it into the energy rich molecule pyruvate. Inside the overall reaction there lie many steps that need to be followed in order for the original glucose molecule to be transformed into pyruvate. The glucose first gathers a phosphate group from an ATP molecule in order to become glucose-6-phosphate. It is then changed into fructose 6-phosphate, with the assistance of phosphoglucose isomerase, which is then changed into fructose 1,6-biphosphate when the fructose molecule receives a phosphate group from another ATP. The next step in the chain is crucial for cells in order to make more energy than they expend through the process of glycolysis; this step is when the fructose 1,6-bisphosphate molecule breaks down into two molecules of dihydroxyacetone phosphate (DHAP), so from this point on whenever ATP is being generated from ADP there are really two ATP molecules generated because there are two molecules undergoing the same reaction.[13] One molecule that takes advantage of this second part of the metabolic process is the fatty molecule glycerol. This is unfortunately prevented if someone is experiencing Glycerol Kinase Deficiency.
When a human's body needs to use stored fat for energy it will release glycerol and other fatty acids into the bloodstream. However, these glycerol molecules must contribute to the process of glycolysis before they can provide energy to the body, as they do not hold the necessary energy within themselves. So before glycerol can enter the pathway of glycolysis it must be converted into an intermediate molecule, which in this case is dihydroxyacetone phosphate (DHAP). This is where glycerol kinase comes into the picture. The enzyme is used in the first step in turning glycerol into dihydroxyacetone phosphate (DHAP). It catalyzes the transfer of a phosphate group from an ATP to a glycerol molecule forming glycerol (3) phosphate. Then glycerol 3-phosphate, with the assistance of glycerol 3-phosphate dehydrogenase, can be dehydrogenated into DHAP. This molecule can then enter the metabolic pathway of glycolysis and provide more energy for the cell.[14] Looking at the entire glycolysis pathway this conversion would yield an extra ATP for each glycerol molecule that eventually made its way into a DHAP molecule, which demonstrates the benefit of releasing glycerol into the bloodstream. However, when suffering from a glycerol kinase deficiency many of the glycerol molecules released into the bloodstream end up not being converted to dihydroxyacetone phosphate (DHAP), because the host does not have enough of the enzyme to catalyze all of the reactions waiting to occur. These extra molecules of glycerol are left floating around in the cell and can cause serious damage if left untreated.
Diagnosis
Classification
GKD can be divided into three distinct forms: infantile, juvenile, and adult. Out of all of these the infantile form is the most clinically relevant because it leads to developmental delay and adrenal insufficiency.[11]
- The infantile form is referred to as complex GKD because the defect in the gene for the glycerol kinase enzyme is interconnected with defects in one or both of its affected genes that are responsible for Duchenne muscular dystrophy and adrenal hypoplasia congenita.[1] The cause of this form is deletion of the Xp21 gene on the X chromosome. Patients have increased levels of serum creatine phosphokinase, which leads to myopathic changes in muscle biopsies resembling Duchenne muscular dystrophy.
- The juvenile form does not show myopathy and patients have normal adrenal function. The main cause leading to this form of GKD is isolated enzyme deficiency.
- The adult form is also caused by an isolated enzyme deficiency. Patients having this form of the disorder are clinically normal.[11]
Treatment
Treatments for Glycerol Kinase Deficiency are targeted to treat the symptoms because there are no permanent treatments for this disease. The main way to treat these symptoms is by using corticosteroids, glucose infusion, or mineralocorticoids. Corticosteroids are steroid hormones that are naturally produced in the adrenal glands. These hormones regulate stress responses, carbohydrate metabolism, blood electrolyte levels, as well as other uses. The mineralocorticoids, such as aldosterone control many electrolyte levels and allow the kidneys to retain sodium. Glucose infusion is coupled with insulin infusion to monitor blood glucose levels and keep them stable.[15]
Due to the multitude of varying symptoms of this disease, there is no specific treatment that will cure this disease altogether. The symptoms can be treated with many different treatments and combinations of medicines to try to find the correct combination to offset the specific symptoms. Everyone with Glycerol Kinase Deficiency has varying degrees of symptoms and thereby requires different medicines to be used in combination to treat the symptoms; however, this disease is not curable and the symptoms can only be managed, not treated fully.[16]
References
- 1 2 Office of Rare Diseases, National Institutes of Health (25 January 2005). "Annual Report on the Rare Diseases and Conditions Research Activities of the National Institutes of Health 1997" (pdf). Retrieved 21 December 2012.
- ↑ Sjarif, D. R.; Hellerud, C.; Van Amstel, J. K. P. V.; Kleijer, W. J.; Sperl, W.; Lacombe, D.; Sass, J. R. O.; Beemer, F. A.; Duran, M.; Poll-The, B. T. (2004). "Glycerol kinase deficiency: Residual activity explained by reduced transcription and enzyme conformation". European Journal of Human Genetics. 12 (6): 424–432. doi:10.1038/sj.ejhg.5201172. PMID 15026783.
- 1 2 Wishart, David (6 March 2010). "Glycerol Kinase Deficiency". Small Molecule Pathway Database. Archived from the original on 2 July 2012. Retrieved 21 December 2012.
- ↑ "Hyperglycerolemia". Online Mendelian Inheritance in Man. National Center for Biotechnology Information. 29 June 2010. Retrieved 21 December 2012.
- 1 2 "Chromosome Xp21 Deletion Syndrome". Online Mendelian Inheritance in Man. National Center for Biotechnology Information. 8 December 2010. Retrieved 21 December 2012.
- ↑ Seltzer, W. K.; Firminger, H.; Klein, J.; Pike, A.; Fennessey, P.; McCabe, E. R. B. (1985). "Adrenal dysfunction in glycerol kinase deficiency". Biochemical Medicine. 33 (2): 189–199. doi:10.1016/0006-2944(85)90027-4. PMID 2988520.
- ↑ Galifer, R. B.; Kalfa, N.; Guibal, M. P. (2004). "What a hidden testicle can hide?...or the clinical traps of cryptorchidism". Archives de Pédiatrie. 11 (4): 350–359. doi:10.1016/j.arcped.2003.11.015. PMID 15139321.
- ↑ Mahbubul Huq, A. H. M.; Lovell, R. S.; Ou, C. -N.; Beaudet, A. L.; Craigen, W. J. (1997). "X-Linked Glycerol Kinase Deficiency in the Mouse Leads to Growth Retardation, Altered Fat Metabolism, Autonomous Glucocorticoid Secretion and Neonatal Death". Human Molecular Genetics. 6 (11): 1803–1809. doi:10.1093/hmg/6.11.1803. PMID 9302256.
- ↑ Rahib, L.; MacLennan, N. K.; Horvath, S.; Liao, J. C.; Dipple, K. M. (2007). "Glycerol kinase deficiency alters expression of genes involved in lipid metabolism, carbohydrate metabolism, and insulin signaling". European Journal of Human Genetics. 15 (6): 646–657. doi:10.1038/sj.ejhg.5201801. PMID 17406644.
- ↑ National Library of Medicine (17 December 2012). "Genotype-Phenotype Correlation". Genetics Home Reference. Retrieved 21 December 2012.
- 1 2 3 Francke, U.; Harper, J. F.; Darras, B. T.; Cowan, J. M.; McCabe, E. R.; Kohlschütter, A.; Seltzer, W. K.; Saito, F.; Goto, J.; Harpey, J. P.; Wise, J. E. (1987). "Congenital adrenal hypoplasia, myopathy, and glycerol kinase deficiency: Molecular genetic evidence for deletions". American Journal of Human Genetics. 40 (3): 212–227. PMC 1684111. PMID 2883886.
- ↑ Richards, Julia E.; Hawley, R. Scott (2010). The Human Genome: A User's Guide. Academic Press. ISBN 978-0-12-333445-9.
- ↑ Benson, Darik (14 August 2012). "Glycolysis". UC Davis ChemWiki. Retrieved 21 December 2012., . "." . University California Davis, 2 Nov. 2010. Web. <>
- ↑ Medh, Jheem D. (August 2006). "Glycolysis" (PDF). Retrieved 21 December 2012.
- ↑ Blau, Nenad; Hoffmann, Georg F.; Leonard, James; Clarke, Joe T.R. (2005). Physician's Guide to the Treatment and Follow-Up of Metabolic Diseases. Springer. ISBN 978-3540229544.
- ↑ "Glycerol Kinase Deficiency". MIC - Metabolic & Genetic Information Centre. 17 October 2010. Retrieved 21 December 2012.