The Barton reaction, also known as the Barton nitrite ester reaction, is a photochemical reaction that involves the photolysis of an alkyl nitrite to form a δ-nitroso alcohol.
Discovered in 1960, the reaction is named for its discoverer, Nobel laureate Sir Derek Barton.[1] Barton's Nobel Prize in Chemistry in 1969 was awarded for his work on understanding conformations of organic molecules, work which was key to realizing the utility of the Barton Reaction.[2]
The Barton reaction involves a homolytic RO–NO cleavage, followed by δ-hydrogen abstraction, free radical recombination, and tautomerization to form an oxime.[3] Selectivity for the δ-hydrogen is a result of the conformation of the 6-membered radical intermediate. Often, the site of hydrogen atom abstraction can be easily predicted. This allows the regio- and stereo-selective introduction of functionality into complicated molecules with high yield. Due to its unique property at the time to change otherwise inert substrates, Barton used this reaction extensively in the 1960s to create a number of unnatural steroid analogues.[4]
While the Barton reaction has not enjoyed the popularity or widespread use of many other organic reactions, together with the mechanistically similar Hofmann–Löffler reaction it represents one of the first examples of C-H activation chemistry, a field which is now the topic of much frontline research in industrial and academic chemistry circles.[5]
Preparation of alkyl nitrites
The unusual alkyl nitrite starting material of the Barton reaction is prepared by attack of an alcohol on a nitrosylium cation generated in situ by dehydration of doubly protonated nitrous acid.[6] This series of steps is mechanistically identical to the first half of the mechanism formation of the more well-known aryl and alkyl diazonium salts.
While the synthesis of alkyl nitrites from nitrosyl chloride is known and oft-employed in the context of complex molecule synthesis, the reaction is reversible and the products are in thermodynamic equilibrium with the starting material. Furthermore, nitrosyl chloride is a powerful oxidizing agent, and oxidation of the alcohols with concomitant chlorination has been observed.[7] The reaction of nitrosyl chloride with aromatic alcohols generally yields nitroso compounds and other over-oxidation products.
Reaction mechanism and regioselectivity
The Barton reaction commences with a photochemically induced cleavage of the nitrite O-N bond, typically using a high pressure mercury lamp.[8] This produces an alkyoxyl radical which immediately abstracts a hydrogen atom from the δ-carbon. In the absence of other radical sources or other proximal reactive groups, the alkyl radical recombines with the nitrosyl radical. The resultant nitroso compounds undergoes tautomerization to the isolated oxime product.

The carbon centered radical can be intercepted by other radical sources such as iodine or acrylonitrile. The first instance results in the δ-hydrogen being replaced with iodine, then subsequent cyclization to a tetrahydrofuran by an SN2 reaction.[9] The second example results in a chain elongation product with the oxime formed 2 carbon units further from the oxygen than normal.[10]
This mechanistic hypothesis is supported by kinetic isotope effect experiments.[11] Isotopic labeling of the nitrite with 15N has shown the mechanism non-‘caged’ and that the nitrosyl radical formed from a given nitrite recombines randomly with other alkyl radicals. However, recombination of the nitrosyl radical with the alkoxyl radical (a reversal of the homolytic cleavage) has been shown to proceed without scrambling of isotope labels.[12] This lack of tight radical pairing is also supported by the observation that alkyl radicals generated by Barton conditions can undergo radical cyclization while analogous intermediates generated by lead tetraacetate oxidation do not.[13]
In rare cases, it appears that the alkoxyl radical may epimerize before hydrogen atom abstraction.[14]
Most commonly, including steroidal systems, the hydrogen atom is abstracted from a methyl group that has a 1,3 diaxial relationship with the alkoxyl radical.[15] In the absence of a hydrogen on the δ-carbon, or when the particular conformation of the substrate orients the ε-carbon close together, 1,6-hydrogen atom transfer is the favored process. However, these reactions tend to be an order of magnitude slower than the corresponding 1,5-hydrogen atom transfer.
Computational studies have shown that this preference for 1,5-hydrogen atom transfer over 1,6-hydrogen atom transfer appears to be entropically favored rather than a result of a particular stable ‘chair-like’ transition state.[16] In fact, it has been calculated that the 1,6-hydrogen atom transfer proceeds through a transition that is about 0.8 kcal/mol lower than that of the 1,5.
In acyclic systems, δ-hydrogen abstraction is still observed, however, alpha-hydrogen abstraction to form the corresponding ketone competes.[17]
In certain cases, particularly nitrites derived from cyclopentyl alcohols, the oxygen-centered radical prefers to react via C-C bond cleavage as opposed to H-atom abstraction.[9] For example, when subjected to Barton conditions, cyclopentyl nitrite forms glutaraldehyde monoxime. This is also observed in cases where the radical intermediate formed by fragmentation is particularly stable, such as the allylic radical formed by the fragmentation of isopulegol nitrite.[18]
Variants
In rigid systems such as aldosterone, the 1,5-hydrogen atom transfer is exceedingly fast, with a rate constant on the order of 10^7 s-1. Similar intermolecular H-atom transfer can be up to 100 times slower.[19] Furthermore, the hydrogen atom transfer benefits from the formation of a stronger O-H bond at the expense of a weaker C-H bond. For the formation of a primary, second, or tertiary alkyl radical from an alkoxyl radical, there is a driving force of 3 kcal/mol, 5 kcal/mol, and 9 kcal/mol, respectively.[15]
The alkyl radical formed after hydrogen atom transfer is susceptible to standard radical reactions when scavengers are present in sufficient excess to outcompete the nitrosyl radical. Soon after their initial disclosure, Barton and co-workers reported the trapping of the radical with I2 and CCl3Br (as Iodine and Bromine radical sources, respectively) to form the δ-halo-alcohol. These halohydrin species can be cyclized to the corresponding tetrahydropyran derivates under basic conditions.[20]
Large excesses of activated alkenes can be used to intercept the alkyl radical and results in formation of a C-C bond from an unactivated C-H bond.[21]
In the presence of oxygen, the alkyl radical is trapped and forms an organic peroxy radical. This intermediate is trapped by the nitrosyl radical and then isomerizes to give a δ-nitrate ester which, while both acid- and base-stable, can be reduced to the corresponding alcohol under mild conditions.[22]
Applications in complex molecule synthesis
Aldosterone acetate
In a publication immediately proceeding Barton's initial disclosure of the methodology in the Journal of the American Chemical Society, a synthesis of aldosterone acetate is demonstrated.[23] Allowing corticosterone acetate to react with nitrosyl chloride in dry pyridine yields the nitrite. Subsequently, irradiation under inert atmosphere followed by treatment with aqueous sodium nitrite selectively gives the desired oxime. The oxime is then acetylated and hydrolyzed to yield the natural product hemiacetal.

Perhydrohistrionicotoxin
After a short synthesis to obtain the desired spiro-[5.4] system, Nobel laureaute E.J. Corey and co-workers employed a Barton reaction to selectively introduce an oxime in a 1,3-diaxial position to the nitrite ester. The oxime is converted to a lactam via a Beckmann rearrangement and then reduced to the natural product.[24]

Azadiradione
Corey again employed the Barton reaction in the synthesis of Azadiradione, a member of the limonoid family of natural products. In this case, nitrosylsulfuric acid is used in place of nitrosyl chloride.[25]

Allobetulin derivatives
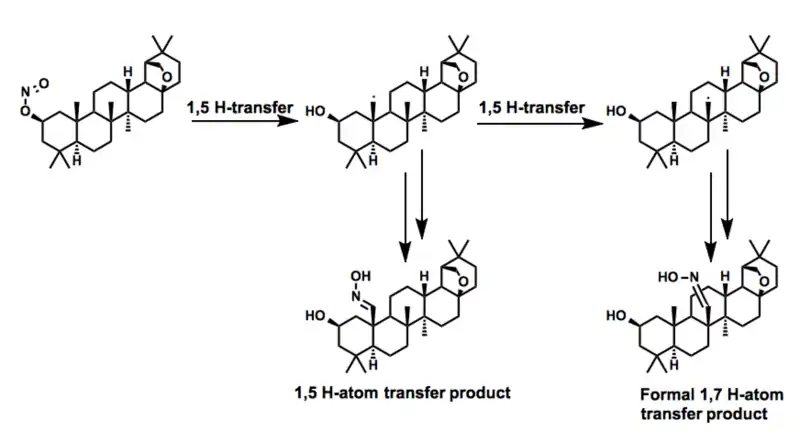
In the process of preparing a series of derivatives of the triterpenoid allobetulin, Dehan and coworkers observed a remarkable transformation resulting from two consecutive 1,5-hydrogen atom transfers. While the product of the single 1,5-hydrogen atom transfer was also observed, the former transformation represent a formal 1,7-hydrogen atom transfer across an enormous distance.[26]
References
- ↑ Barton, D. H. R.; Beaton, J. M.; Geller, L. E.; Pechet, M. M. (1960). "A New Photochemical Reaction". Journal of the American Chemical Society. 82 (10): 2640–2641. doi:10.1021/ja01495a061.
- ↑ Barton, D. H. R.; Beaton, J. M.; Geller, L. E.; Pechet, M. M. (1961). "A New Photochemical Reaction1". Journal of the American Chemical Society. 83 (19): 4076–4083. doi:10.1021/ja01480a030.
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Barton Reaction". doi:10.1351/goldbook.B00599
- ↑ Nussbaum, A. L.; Yuan, E. P.; Robinson, C. H.; Mitchell, A.; Oliveto, E. P.; Beaton, J. M.; Barton, D. H. R. (1962). "The Photolysis of Organic Nitrites. VII. Fragmentation of the Steroidal Side Chain". The Journal of Organic Chemistry. 27: 20–23. doi:10.1021/jo01048a004.
- ↑ Gutekunst, W. R.; Baran, P. S. (2011). "C–H functionalization logic in total synthesis". Chemical Society Reviews. 40 (4): 1976–91. doi:10.1039/c0cs00182a. PMID 21298176.
- ↑ "N-Butyl Nitrite". Organic Syntheses. 16: 7. 1936. doi:10.15227/orgsyn.016.0007.
- ↑ Beckham, L. J.; Fessler, W. A.; Kise, M. A. (1951). "Nitrosyl Chloride". Chemical Reviews. 48 (3): 319–396. doi:10.1021/cr60151a001. PMID 24541207.
- ↑ Sugimoto, A.; Fukuyama, T.; Sumino, Y.; Takagi, M.; Ryu, I. (2009). "Microflow photo-radical reaction using a compact light source: Application to the Barton reaction leading to a key intermediate for myriceric acid A". Tetrahedron. 65 (8): 1593–1598. doi:10.1016/j.tet.2008.12.063.
- 1 2 Akhtar, M.; Barton, D. H. R.; Sammes, P. G. (1965). "Some Radical Exchange Reactions during Nitrite Ester Photolysis1". Journal of the American Chemical Society. 87 (20): 4601–4607. doi:10.1021/ja00948a036.
- ↑ Petrovic, G.; Cekovic, Z. (1997). "Free radical alkylation of the remote nonactivated δ-carbon atom". Tetrahedron Lett. 38 (4): 627–630. doi:10.1016/s0040-4039(96)02357-x.
- ↑ Barton, D. H. R.; Hesse, R. H.; Pechet, M. M.; Smith, L. C. (1979). "The mechanism of the barton reaction". Journal of the Chemical Society, Perkin Transactions 1: 1159. doi:10.1039/P19790001159. hdl:1893/35137.
- ↑ Akhtar, M.; Pechet, M. M. (1964). "The Mechanism of the Barton Reaction". Journal of the American Chemical Society. 86 (2): 265–268. doi:10.1021/ja01056a035.
- ↑ Čeković, Ẑ.; Ilijev, D. (1988). "Intramolecular cyclization of alkenyl radicals generated by 1,5-hydrogen transfer to alkoxy radicals". Tetrahedron Letters. 29 (12): 1441–1444. doi:10.1016/S0040-4039(00)80319-6.
- ↑ Nickson, A.; Mahajan, J.; McGuire, F. (1961). "Communications- Epimerization in a Nitrite Ester Photolysis". The Journal of Organic Chemistry. 26 (9): 3617–3618. doi:10.1021/jo01067a671.
- 1 2 Čeković, Ž. (2003). "Reactions of δ-carbon radicals generated by 1,5-hydrogen transfer to alkoxyl radicals". Tetrahedron. 59 (41): 8073–8090. doi:10.1016/S0040-4020(03)01202-X.
- ↑ Dorigo, A. E.; McCarrick, M. A.; Loncharich, R. J.; Houk, K. N. (1990). "Transition structures for hydrogen atom transfers to oxygen. Comparisons of intermolecular and intramolecular processes, and open- and closed-shell systems". Journal of the American Chemical Society. 112 (21): 7508–7514. doi:10.1021/ja00177a009.
- ↑ Ishmuratov, G. Y.; Kharisov, R. Y.; Shayakhmetova, A. K.; Botsman, L. P.; Shitikova, O. V.; Tolstikov, G. A. (2005). "Ozonolysis of Ricinolic Acid Derivatives and Transformations of the Ozonolysis Products under Barton Reaction Conditions". Chemistry of Natural Compounds. 41 (6): 643–649. doi:10.1007/s10600-006-0003-z. S2CID 43171151.
- ↑ Bulliard, M.; Balme, G. V.; Gore, J. (1989). "Fragmentation of isopulegol by a radical process". Tetrahedron Letters. 30 (17): 2213–2216. doi:10.1016/S0040-4039(00)99651-5.
- ↑ Robertson, J.; Pillai, J.; Lush, R. K. (2001). "Radical translocation reactions in synthesis". Chemical Society Reviews. 30 (2): 94–103. doi:10.1039/b000705f.
- ↑ Akhtar, M.; Barton, D. H. R.; Sammes, P. G. (1964). "Radical Exchange during Nitrite Photolysis". Journal of the American Chemical Society. 86 (16): 3394–3395. doi:10.1021/ja01070a039.
- ↑ Petrović, G.; Čeković, Ž. (1999). "Alkylation of remote non-activated δ-carbon atoms: Addition of δ-carbon radicals, generated by 1,5-hydrogen transfer in alkoxy radical intermediates, to activated olefins". Tetrahedron. 55 (5): 1377–1390. doi:10.1016/S0040-4020(98)01110-7.
- ↑ Allen, J.; Boar, R. B.; McGhie, J. F.; Barton, D. H. R. (1973). "Nitrite photolysis in the presence of oxygen. An improved synthesis of 32-oxygenated lanostanes". Journal of the Chemical Society, Perkin Transactions 1: 2402. doi:10.1039/P19730002402.
- ↑ Barton, D. H. R.; Beaton, J. M. (1960). "A Synthesis of Aldosterone Acetate". Journal of the American Chemical Society. 82 (10): 2641. doi:10.1021/ja01495a062.
- ↑ Corey, E. J. (1975). "Simple total synthesis of (+-)-perhydrohistrionicotoxin". Journal of the American Chemical Society. 97 (2): 430–431. doi:10.1021/ja00835a039. PMID 1169269.
- ↑ Corey, E. J.; Hahl, R. W. (1989). "Synthesis of a limonoid, azadiradione". Tetrahedron Letters. 30 (23): 3023–3026. doi:10.1016/S0040-4039(00)99392-4.
- ↑ Dehaen, W.; Mashentseva, A. A.; Seitembetov, T. S. (2011). "Allobetulin and Its Derivatives: Synthesis and Biological Activity". Molecules. 16 (12): 2443–2466. doi:10.3390/molecules16032443. PMC 6259842. PMID 21403601.
- László Kürti, Barbara Czakó: Strategic Applications of Named Reactions in Organic Synthesis; Elsevier Academic Press, Burlington-San Diego-London 2005, 1. Edition; ISBN 0-12-369483-3.