In chemistry, the selection rule (also known as the transition rule) formally restricts certain reactions, known as spin-forbidden reactions, from occurring due to a required change between two differing quantum states. When a reactant exists in one spin state and the product exists in a different spin state, the corresponding reaction will have an increased activation energy when compared to a similar reaction in which the spin states of the reactant and product are isomorphic. As a result of this increased activation energy, a decreased rate of reaction is observed.
Case of some cobalt carbonyls

Singlet and triplet states can occur within organometallic complexes as well, such as Tpi-Pr,MeCo(CO)2 and Tpi-Pr,MeCo(CO), respectively.
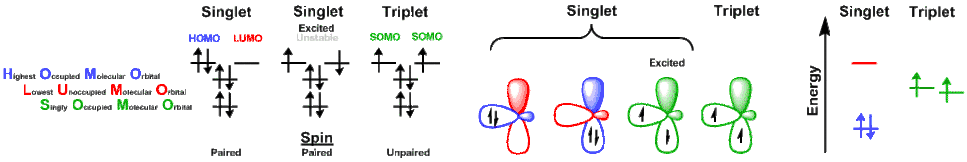
Changing spin states
When a reaction converts a metal from a singlet to triplet state (or vice versa):
- The energy of the two spin states must be nearly equal, as dictated by temperature,
- A mechanism is required to change spin states.
Strong spin-orbital coupling can satisfy the 2nd condition. Parameter 1, however, can lead to very slow reactions due to large disparities between the metal complex's potential energy surfaces, which only cross at high energy leading to a substantial activation barrier.[2]
Spin-forbidden reactions formally fall into the category of electronically non-adiabatic reactions.[3] In general, potential energy surfaces fall into either the adiabatic and diabatic classification. Potential Energy Surfaces that are adiabatic rely on the use of the full electronic Hamiltonian, which includes the spin-orbit term. Those that are diabatic are likewise derived by solving the eigenvalues of the Schrödinger equation, but in this case one or more terms are omitted.[4]
Non-adiabatic transition

Once a minimum energy crossing point is reached and parameter 1 above is satisfied, the system needs to hop from one diabatic surface to the other, as stated above by parameter 2. At a given energy (E), the rate coefficient [k(E)] of a spin-forbidden reaction can be calculated using the density of rovibrational states of the reactant [ρ(E)] and the effective integrated density of states in the crossing seam between the two surfaces [Ner(E)].

where

The probability of hopping (psh) is calculated from Landau-Zener theory giving

where

in which the spin-orbit coupling derived off the diagonal Hamiltonian matrix element between two electronic states (H12), the relative slope of the two surfaces at the crossing seam [F(Δ)], the reduced mass of the system through its movement along the hopping coordinate (μ), and the kinetic energy of the system passing through the crossing point (E) are used.
It is useful to note that when Eh < Ec (when below the minimum energy crossing point) the probability of hopping between spin states is null.[5]
An example of a spin-forbidden reaction

One example showing the slowing effect of spin-forbidden reaction takes place when Fe(CO)x is placed under CO pressure. Transitions from x = 2,3,4 to x = 3,4,5 demonstrate a slowing rate when the reactant is in a triplet ground state but the product is in a singlet ground state. In the case of Fe(CO)x, when x = 2, 3, 4 the iron exists as a triplet in ground state; when x = 5, the iron exists as a singlet in ground state. The kinetics of said system can be represented by:
![{\displaystyle {\ce {Fe(CO)2->[{k_{23}}][CO]Fe(CO)3->[{k_{34}}][CO]Fe(CO)4->[{k_{45}}][CO]Fe(CO)5}}}](../I/5bba6aa1ab4092d76cdd505c9d0d7dcac1f3b13e.svg)
where:
- ,
- , and
- .



The rate constants above are notably temperature independent, tested at 55 °C, 21 °C, and 10 °C, indicating, according to the authors, that the observed 500 fold reduction in rate occurs due to the spin-forbidden nature of the latter equation and not due to the kinetics of adding an additional ligand.[7]
Application to catalysis
Ligand association and dissociation from metal centers involve a fundamental change in the coordination sphere of that metal. This change in certain reactions also requires a change in spin state to occur, which can retard the rate of ligand association. Oxidative addition and reductive elimination can be thought of in a similar manner to ligand association and dissociation, respectively.[8] Mathematically, the rate ligand dissociation can increase when the reaction proceeds from one spin state to another, although it must be noted that this effect is often small in comparison with other factors such as sterics around the metal center.[9][10]
C-H activation
Insertion into C-H bonds, known as C-H activation, is an integral first step in C-H functionalization.[11] For some metal complexes with identical ligands, C-H activation is rapid when one metal is used and slow when other metals are used, often first row transition metals, due to the spin allowed nature of the former case and the spin-forbidden nature of the latter case. The difference in rates of C-H activation of methane for CoCp(CO), RhCp(CO), and IrCp(CO) readily demonstrate this property. CoCp(CO), the starting material in a C-H activation, exists in a triplet spin state while RhCp(CO) exists in a singlet state, with the triplet state only 5.9 kcal/mol away. IrCp(CO) is unique among these complexes in that its starting state is essentially degenerate between the triplet and singlet states. The given product of C-H insertion, CpMH(CO)(CH3), where M = Co, Rh, Ir, is in a singlet state meaning that the C-H activation with CoCp(CO) must reach the minimum energy crossing point for the reactant and product's potential energy surfaces, thus requiring relatively high energies to proceed.[12]

Oxidative addition into silicon-hydrogen bonds
Through the use of photolysis, the rate of oxidative addition into silicon-hydrogen bonds has been shown to increase when the starting material is excited to the correct spin-state. CpRe(CO)2 and CpMn(CO)2 were subjected to photolysis to change their spin states from triplet to singlet and vice versa, respectively, such that an oxidative addition to Et3Si-H could occur at a greatly accelerated rate.[13]

Oxidation chemistry

Metal-oxo species, due to their small spatial extent of metal-centered d orbitals leading to weak bonding, often have similar energies for both the low spin () and high spin configuration ().[14] This similarity in energy between the low- and high spin configurations of oxo-species lends itself to the study of spin-forbidden reactions, such as Mn(salen)-catalyzed epoxidation. The Mn(salen)-oxo species can exist in either a triplet or quintet state. While the product of the quintet lies at a lower energy, both the triplet and quintet products can be observed.[15]



References
- ↑ Theopold, Klaus H (1995). "Can Spin State Change Slow Organometallic Reactions". Journal of the American Chemical Society. 117 (47): 11745–8. doi:10.1021/ja00152a015.
- ↑ Cundari, Thomas (2001). Computational Organometallic Chemistry. Marcel Dekker, Inc. pp. 293. ISBN 9780824704780.
- ↑ Cundari, Thomas (2001). Computational Organometallic Chemistry. Marcel Dekker, Inc. pp. 294. ISBN 9780824704780.
- ↑ Harvey, Jeremy (2006). "Understanding the Kinetics of Spin-Forbidden Chemical Reactions". Physical Chemistry Chemical Physics. 9 (3): 331–2. doi:10.1039/b614390c. PMID 17199148.
- ↑ Harvey, Jeremy (2006). "Understanding the Kinetics of Spin-Forbidden Chemical Reactions". Physical Chemistry Chemical Physics. 9 (3): 332–3. doi:10.1039/b614390c. PMID 17199148.
- ↑ Kirchner, Karl (2010). "Reactivity of coordinatively unsaturated iron complexes towards carbon monoxide: to bind or not to bind?". Dalton Transactions. 40 (18): 4778–4792. doi:10.1039/c0dt01636e. PMID 21380474.
- ↑ Weitz, Eric (1986). "The wavelength dependence of excimer laser photolysis of Fe(CO)5 in the gas phase. Transient infrared spectroscopy and kinetics of the FeCOx (x=4,3,2) photofragments". The Journal of Chemical Physics. 84 (4): 1977–1986. Bibcode:1986JChPh..85.1977S. doi:10.1063/1.451141.
- ↑ Cundari, Thomas (2001). Computational Organometallic Chemistry. Marcel Dekker, Inc. pp. 299–303. ISBN 9780824704780.
- ↑ Poli, Rinaldo (1995). "Dissociative phosphine exchange for cyclopentadienylmolybdenum(III) systems. Bridging the gap between Werner-like coordination chemistry and low-valent organometallic chemistry" (PDF). Inorganica Chimica Acta. 240 (1–2): 355–66. doi:10.1016/0020-1693(95)04554-6.
- ↑ Poli, Rinaldo (1996). "Molybdenum Open-Shell Organometallics. Spin State Changes in Pairing Energy Effects" (PDF). Journal of the American Chemical Society. 30 (12): 494–501. doi:10.1021/ar960280g.
- ↑ Organometallic C–H Bond Activation: An Introduction Alan S. Goldman and Karen I. Goldberg ACS Symposium Series 885, Activation and Functionalization of C–H Bonds, 2004, 1–43
- ↑ Siegbahn, Per (1996). "Comparison of the C-H Activation of Methane by M(C5H5)(CO) for M=Cobalt, Rhodium, and Iridium". Journal of the American Chemical Society. 118 (6): 1487–96. doi:10.1021/ja952338c.
- ↑ Harris, Charles (1999). "Ultrafast Infrared Studies of Bond Activation in Organometallic Complexes". Acc. Chem. Res. 32 (7): 551–60. doi:10.1021/ar970133y.
- ↑ Cudari, Thomas (2001). Computational Organometallic Chemistry. Marcel Dekker Inc. pp. 301–2.
- ↑ Linde, C.; Åkermark, B.; Norrby, P.-O.; Svensson, M. (1999). "Timing Is Critical: Effect of Spin Changes on the Diastereoslectivity in Mn(salen)-Catalyzed Epoxidation". Journal of the American Chemical Society. 121 (21): 5083–4. doi:10.1021/ja9809915.