RESOLFT, an acronym for REversible Saturable OpticaL Fluorescence Transitions, denotes a group of optical fluorescence microscopy techniques with very high resolution. Using standard far field visible light optics a resolution far below the diffraction limit down to molecular scales can be obtained.
With conventional microscopy techniques, it is not possible to distinguish features that are located at distances less than about half the wavelength used (i.e. about 200 nm for visible light). This diffraction limit is based on the wave nature of light. In conventional microscopes the limit is determined by the used wavelength and the numerical aperture of the optical system. The RESOLFT concept surmounts this limit by temporarily switching the molecules to a state in which they cannot send a (fluorescence-) signal upon illumination. This concept is different from for example electron microscopy where instead the used wavelength is much smaller.
Working principle
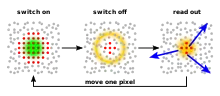
RESOLFT microscopy is an optical microscopy with very high resolution that can image details in samples that cannot be imaged with conventional or confocal microscopy. Within RESOLFT the principles of STED microscopy[1][2] and GSD microscopy are generalized. Structures that are normally too close to each other to be distinguished are read out sequentially.
Within this framework all methods can be explained that operate on molecules that have at least two distinguishable states, where reversible switching between the two states is possible, and where at least one such transition can be optically induced.
In most cases fluorescent markers are used, where one state (A) which is bright, that is, generates a fluorescence signal, and the other state (B) is dark, and gives no signal. One transition between them can be induced by light (e.g. A→B, bright to dark).
The sample is illuminated inhomogeneously with the illumination intensity at one position being very small (zero under ideal conditions). Only at this place are the molecules never in the dark state B (if A is the pre-existing state) and remain fully in the bright state A. The area where molecules are mostly in the bright state can be made very small (smaller than the conventional diffraction limit) by increasing the transition light intensity (see below). Any signal detected is thus known to come only from molecules in the small area around the illumination intensity minimum. A high resolution image can be constructed by scanning the sample, i.e., shifting the illumination profile across the surface.[3]
The transition back from B to A can be either spontaneous or driven by light of another wavelength. The molecules have to be switchable several times in order to be present in state A or B at different times during scanning the sample. The method also works if the bright and the dark state are reversed, one then obtains a negative image.
Resolution below the diffraction-limit

In RESOLFT the area where molecules reside in state A (bright state) can be made arbitrarily small despite the diffraction-limit.
- One has to illuminate the sample inhomogeneously so that an isolated zero intensity point is created. This can be achieved e.g. by interference.
- At low intensities (lower than the blue line in the image) most marker molecules are in the bright state, if the intensity is above, most markers are in the dark state.
Upon weak illumination we see that the area where molecules remain in state A is still quite large because the illumination is so low that most molecules reside in state A. The shape of the illumination profile does not need to be altered. Increasing the illumination brightness already results in a smaller area where the intensity is below the amount for efficient switching to the dark state. Consequently, also the area where molecules can reside in state A is diminished. The (fluorescence) signal during a following readout originates from a very small spot and one can obtain very sharp images.
In the RESOLFT concept, the resolution can be approximated by , whereby is the characteristic intensity required for saturating the transition (half of the molecules remain in state A and half in state B), and denotes the intensity applied. If the minima are produced by focusing optics with a numerical aperture , the minimal distance at which two identical objects can be discerned is which can be regarded as an extension of Abbe’s equation. The diffraction-unlimited nature of the RESOLFT family of concepts is reflected by the fact that the minimal resolvable distance can be continuously decreased by increasing . Hence the quest for nanoscale resolution comes down to maximizing this quantity. This is possible by increasing or by lowering .







Variants
Different processes are used when switching the molecular states. However, all have in common that at least two distinguishable states are used. Typically the fluorescence property used marks the distinction of the states, however this is not essential, as absorption or scattering properties could also be exploited.[4]
STED Microscopy
(Main article STED microscopy)
Within the STED microscopy (STimulated Emission Depletion microscopy)[1][2] a fluorescent dye molecule is driven between its electronic ground state and its excited state while sending out fluorescence photons. This is the standard operation mode in fluorescence microscopy and depicts state A. In state B the dye is permanently kept in its electronic ground state through stimulated emission. If the dye can fluoresce in state A and not in state B, the RESOLFT concept applies.
GSD microscopy
(Main article GSD microscopy)
GSD microscopy (Ground State Depletion microscopy) also uses fluorescent markers. In state A, the molecule can freely be driven between the ground and the first excited state and fluorescence can be sent out. In the dark state B the ground state of the molecule is depopulated, a transition to a long lived excited state takes place from which fluorescence is not emitted. As long as the molecule is in the dark state, it's not available for cycling between ground and excited state, fluorescence is hence turned off.
SPEM and SSIM
SPEM (Saturated Pattern Excitation Microscopy)[5] and SSIM (Saturated Structured Illumination Microscopy)[6] are exploiting the RESOLFT concept using saturated excitation to produce "negative" images, i.e. fluorescence occurs from everywhere except at a very small region around the geometrical focus of the microscope. Also non point-like patterns are used for illumination. Mathematical image reconstruction is necessary to obtain positive images again.
RESOLFT with switchable proteins
Some fluorescent proteins can be switched on and off by light of an appropriate wavelength. They can be used in a RESOLFT-type microscope.[7] During illumination with light, these proteins change their conformation. In the process they gain or lose their ability to emit fluorescence. The fluorescing state corresponds to state A, the non-fluorescing to state B and the RESOLFT concept applies again. The reversible transition (e.g. from B back to A) takes place either spontaneously or again driven by light. Inducing conformational changes in proteins can be achieved already at much lower switching light intensities as compared to stimulated emission or ground state depletion (some W/cm²). In combination with 4Pi microscopy images with isotropic resolution below 40 nm have been taken of living cells at low light levels.[8]
RESOLFT with switchable organic dyes
Just as with proteins, also some organic dyes can change their structure upon illumination.[9][10] The ability to fluoresce of such organic dyes can be turned on and off through visible light. Again the applied light intensities can be quite low (some 100 W/cm²).
References
- 1 2 Stefan W. Hell & Jan Wichmann (1994). "Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy". Optics Letters. 19 (11): 780–2. Bibcode:1994OptL...19..780H. doi:10.1364/OL.19.000780. PMID 19844443.
- 1 2 Thomas A. Klar; Stefan W. Hell (1999). "Subdiffraction resolution in far-field fluorescence microscopy". Optics Letters. 24 (14): 954–956. Bibcode:1999OptL...24..954K. doi:10.1364/OL.24.000954. PMID 18073907.
- ↑ See also Confocal laser scanning microscopy.
- ↑ Stefan W. Hell (2004). "Strategy for far-field optical imaging and writing without diffraction limit". Physics Letters A. 326 (1–2): 140–145. Bibcode:2004PhLA..326..140H. doi:10.1016/j.physleta.2004.03.082.
- ↑ Rainer Heintzmann; Thomas M. Jovin; Christoph Cremer (2002). "Saturated patterned excitation microscopy a concept for optical resolution improvement". Journal of the Optical Society of America A. 19 (8): 1599–2109. Bibcode:2002JOSAA..19.1599H. doi:10.1364/JOSAA.19.001599. hdl:11858/00-001M-0000-0029-2C24-9. PMID 12152701.
- ↑ Mats G. L. Gustafsson (2005). "Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution". Proceedings of the National Academy of Sciences of the United States of America. 102 (37): 13081–6. Bibcode:2005PNAS..10213081G. doi:10.1073/pnas.0406877102. PMC 1201569. PMID 16141335.
- ↑ Michael Hofmann; Christian Eggeling; Stefan Jakobs; Stefan W. Hell (2005). "Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins". Proceedings of the National Academy of Sciences of the United States of America. 102 (49): 17565–9. Bibcode:2005PNAS..10217565H. doi:10.1073/pnas.0506010102. PMC 1308899. PMID 16314572.
- ↑ Ulrike Böhm; Stefan W. Hell; Roman Schmidt (2016). "4Pi-RESOLFT nanoscopy". Nature Communications. 7 (10504): 1–8. Bibcode:2016NatCo...710504B. doi:10.1038/ncomms10504. PMC 4740410. PMID 26833381.
- ↑ Mariano Bossi; Jonas Fölling; Marcus Dyba; Volker Westphal; Stefan W. Hell (2006). "Breaking the diffraction resolution barrier in far-field microscopy by molecular optical bistability". New Journal of Physics. 8 (11): 275. Bibcode:2006NJPh....8..275B. doi:10.1088/1367-2630/8/11/275.
- ↑ Jiwoong Kwon; Jihee Hwang; Jaewan Park; Gi Rim Han; Kyu Young Han; Seong Keun Kim (2015). "RESOLFT nanoscopy with photoswitchable organic fluorophores". Scientific Reports. 5: 17804. Bibcode:2015NatSR...517804K. doi:10.1038/srep17804. PMC 4671063. PMID 26639557.