| Crystal Structure of Prp8 | |||||||
|---|---|---|---|---|---|---|---|
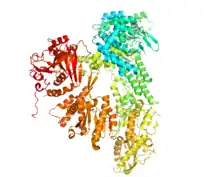 Prp8 Crystal Structure | |||||||
| Identifiers | |||||||
| Symbol | PRP8 | ||||||
| Alt. symbols | USA2, DBF3, DNA39, RNA8, SLT21 | ||||||
| PDB | 4I43 | ||||||
| UniProt | P33334 | ||||||
| |||||||
Prp8 refers to both the Prp8 protein and Prp8 gene. Prp8's name originates from its involvement in pre-mRNA processing. The Prp8 protein is a large, highly conserved, and unique protein that resides in the catalytic core of the spliceosome and has been found to have a central role in molecular rearrangements that occur there. Prp8 protein is a major central component of the catalytic core in the spliceosome, and the spliceosome is responsible for splicing of precursor mRNA that contains introns and exons. Unexpressed introns are removed by the spliceosome complex in order to create a more concise mRNA transcript. Splicing is just one of many different post-transcriptional modifications that mRNA must undergo before translation. Prp8 has also been hypothesized to be a cofactor in RNA catalysis.[1]
History
The systematic name for the PRP8 protein is YHR165C. Prp8 protein is coded by a single gene in humans with 42 exons. The size of Prp8 ranges between 230 and 280 kDa depending on the organism. The sequence coding for the Prp8 protein is highly conserved between eukaryotic organisms, with a 61% identity match between humans and yeast in amino acid sequence.[1] The Prp8 gene is located on chromosome VIII in yeast and chromosome 17 in humans.
Role in splicing
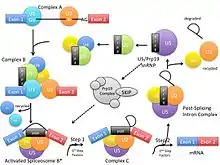
Pre-mRNA splicing involves two trans-esterification reactions and attacks by hydroxyl groups within the spliceosome. In these reactions, spliceosomal intron removal is catalyzed by the spliceosome using the same mechanism as Group II introns.[2] There are five key small nuclear RNA-protein complexes (snRNP) involved in this process. All of the snRNPs together contribute about 50 proteins to the core spliceosome.[2] The Prp8 gene encodes for a protein that is a central part of the U5 snRNP and the U5-U4/U6 tri-snRNP. The U5-U4/U6 tri-snRNP is involved with Complex B, the pre-catalytic spliceosome, where the U5 snRNP binds to exons at the 5’ end of the mRNA before shifting to introns. The U5 snRNP is involved with Complex C, the catalytic spliceosome, where the U5 snRNP binds to an exon at the 3’ splice site and the lariat loop forms. The U5 snRNP is also involved with Complex C*, the post-catalytic spliceosome, where it remains bound to the lariat before the spliced RNA is released and the snRNPs are recycled.
Common research methods for studying the structure and functions of Prp8 are co-immunoprecipitation and Western blot analysis. The structure of Prp8 includes a RNA recognition motif, a MPN / JAB ubiquitin-binding domain near the C-terminus, and a nuclear localization signal (NLS) that tags the protein to be moved to the cell nucleus.[3] The crystal structure of Prp8 protein (residues 885–2413) reveals tightly associated domains that resemble an intron reverse transcriptase and a type II restriction endonuclease. This implies that Prp8 might play roles similar to both the creation of cDNA and in cutting the DNA during splicing.
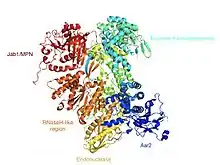
Prp8 is also more involved with maintaining proper conformation of the bound RNA cofactors and substrates of the splicing reaction. Prp8, along with two other U5 snRNP proteins, helps to activate the spliceosome and form its catalytic active center. It has been proposed that GTP hydrolysis results in a rearrangement of Prp8 that releases the U1 and U4 snRNPs and is responsible for this activation of the catalytic core of the spliceosome.[4] Prp8 performs a scaffold-like function in the spliceosome and holds onto many of the interacting substrates and subunits. It has been cross-linked at both the 3’ and 5’ splice sites in mRNA.[5] Due to these structural elements, it has been assumed that Prp8 may have evolved from inactivated retroelements of reverse transcriptases,[6] with the snRNPs replacing the catalytic domains of self-splicing ancestors.[2]
Mutation and disease
Deficiencies
Prp8 mutation has been linked to the human disease Retinitis Pigmentosa causing vision loss, especially progressing into adulthood. This autosomal dominant affliction results with degeneration of the photoreceptors of the retina of the eye. This disorder is caused by mutations in the C-terminus. Retinitis Pigmentosa results from nine missense mutations in the last exon of the mature mRNA result with changes in seven highly conserved amino acids. Studies in yeast indicate that mutation of the C-terminus affects interactions with Brr2p, a helicase responsible for necessary function for the unwinding of the U1 snRNA/5’SS and U4/U6 RNA helices.
Phenotype mutations of Prp8 across species
Caenorhabditis elegans Prp8 has been linked to reproduction and development. RNAi, or RNA Interference, was used to knockout Prp8. This resulted in a high level of sterility, a clear body, and protruding vulva, all phenotypical expressions linked to reproduction and development.[7][8] Mouse Prp8 mutation has resulted in Retinitis Pigmentosa (see above).[9] Yeast Prp8 mutation results in a U5 snRNP maturation defect.[10] The U5 snRNAP component of the splicesosome is necessary to bind to the 5' and 3' exons during pre-mRNA splicing. Mutations with this subunit correlate to reduced or inaccurate editing of RNA. In severe cases, mutations in Prp8 can lead to cell death.
See also
References
- 1 2 Grainger RJ, Beggs JD (May 2005). "Prp8 protein: at the heart of the spliceosome". RNA. 11 (5): 533–57. doi:10.1261/rna.2220705. PMC 1370742. PMID 15840809.
- 1 2 3 Cox DL, Nelson MM (2008). Lehninger Principles of Biochemistry (5th ed.). New York: W.H. Freeman. pp. 1037–1039. ISBN 9780716771081.
- ↑ Bellare P, Kutach AK, Rines AK, Guthrie C, Sontheimer EJ (February 2006). "Ubiquitin binding by a variant Jab1/MPN domain in the essential pre-mRNA splicing factor Prp8p". RNA. 12 (2): 292–302. doi:10.1261/rna.2152306. PMC 1370909. PMID 16428608.
- ↑ Strauss, E.J.; Ben-Yehuda, S.; Umen, J.G.; Wiesner, S.; Brenner, T.J. "PRP8 - U4/U6-U5 snRNP complex component PRP8". WikiGenes. Collaborative Publishing. Retrieved 2 November 2015.
- ↑ Galej WP, Oubridge C, Newman AJ, Nagai K (January 2013). "Crystal structure of Prp8 reveals active site cavity of the spliceosome". Nature. 493 (7434): 638–43. Bibcode:2013Natur.493..638G. doi:10.1038/nature11843. PMC 3672837. PMID 23354046.
- ↑ Dlakić M, Mushegian A (May 2011). "Prp8, the pivotal protein of the spliceosomal catalytic center, evolved from a retroelement-encoded reverse transcriptase". RNA. 17 (5): 799–808. doi:10.1261/rna.2396011. PMC 3078730. PMID 21441348.
- ↑ Gönczy P, Echeverri C, Oegema K, Coulson A, Jones SJ, Copley RR, Duperon J, Oegema J, Brehm M, Cassin E, Hannak E, Kirkham M, Pichler S, Flohrs K, Goessen A, Leidel S, Alleaume AM, Martin C, Ozlü N, Bork P, Hyman AA (November 2000). "Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III". Nature. 408 (6810): 331–6. Bibcode:2000Natur.408..331G. doi:10.1038/35042526. PMID 11099034. S2CID 4364278.
- ↑ McKie AB, McHale JC, Keen TJ, Tarttelin EE, Goliath R, van Lith-Verhoeven JJ, Greenberg J, Ramesar RS, Hoyng CB, Cremers FP, Mackey DA, Bhattacharya SS, Bird AC, Markham AF, Inglehearn CF (July 2001). "Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13)". Human Molecular Genetics. 10 (15): 1555–62. doi:10.1093/hmg/10.15.1555. PMID 11468273.
- ↑ Graziotto JJ, Farkas MH, Bujakowska K, Deramaudt BM, Zhang Q, Nandrot EF, Inglehearn CF, Bhattacharya SS, Pierce EA (January 2011). "Three gene-targeted mouse models of RNA splicing factor RP show late-onset RPE and retinal degeneration". Investigative Ophthalmology & Visual Science. 52 (1): 190–8. doi:10.1167/iovs.10-5194. PMC 3053274. PMID 20811066.
- ↑ Boon KL, Grainger RJ, Ehsani P, Barrass JD, Auchynnikava T, Inglehearn CF, Beggs JD (November 2007). "prp8 mutations that cause human retinitis pigmentosa lead to a U5 snRNP maturation defect in yeast". Nature Structural & Molecular Biology. 14 (11): 1077–83. doi:10.1038/nsmb1303. PMC 2584834. PMID 17934474.