| Part of a series on |
| Evolutionary biology |
|---|
 |
|


A phylogenetic tree, phylogeny or evolutionary tree is a graphical representation which shows the evolutionary history between a set of species or taxa during a specific time.[3][4] In other words, it is a branching diagram or a tree showing the evolutionary relationships among various biological species or other entities based upon similarities and differences in their physical or genetic characteristics. In evolutionary biology, all life on Earth is theoretically part of a single phylogenetic tree, indicating common ancestry. Phylogenetics is the study of phylogenetic trees. The main challenge is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of species or taxa. Computational phylogenetics (also phylogeny inference) focuses on the algorithms involved in finding optimal phylogenetic tree in the phylogenetic landscape.[3][4]
Phylogenetic trees may be rooted or unrooted. In a rooted phylogenetic tree, each node with descendants represents the inferred most recent common ancestor of those descendants,[5] and the edge lengths in some trees may be interpreted as time estimates. Each node is called a taxonomic unit. Internal nodes are generally called hypothetical taxonomic units, as they cannot be directly observed. Trees are useful in fields of biology such as bioinformatics, systematics, and phylogenetics. Unrooted trees illustrate only the relatedness of the leaf nodes and do not require the ancestral root to be known or inferred.
History
The idea of a tree of life arose from ancient notions of a ladder-like progression from lower into higher forms of life (such as in the Great Chain of Being). Early representations of "branching" phylogenetic trees include a "paleontological chart" showing the geological relationships among plants and animals in the book Elementary Geology, by Edward Hitchcock (first edition: 1840).
Charles Darwin featured a diagrammatic evolutionary "tree" in his 1859 book On the Origin of Species. Over a century later, evolutionary biologists still use tree diagrams to depict evolution because such diagrams effectively convey the concept that speciation occurs through the adaptive and semirandom splitting of lineages.
The term phylogenetic, or phylogeny, derives from the two ancient greek words φῦλον (phûlon), meaning "race, lineage", and γένεσις (génesis), meaning "origin, source".[6][7]
Properties
Rooted tree

A rooted phylogenetic tree (see two graphics at top) is a directed tree with a unique node — the root — corresponding to the (usually imputed) most recent common ancestor of all the entities at the leaves of the tree. The root node does not have a parent node, but serves as the parent of all other nodes in the tree. The root is therefore a node of degree 2, while other internal nodes have a minimum degree of 3 (where "degree" here refers to the total number of incoming and outgoing edges).
The most common method for rooting trees is the use of an uncontroversial outgroup—close enough to allow inference from trait data or molecular sequencing, but far enough to be a clear outgroup. Another method is midpoint rooting, or a tree can also be rooted by using a non-stationary substitution model.[8]
Unrooted tree

Unrooted trees illustrate the relatedness of the leaf nodes without making assumptions about ancestry. They do not require the ancestral root to be known or inferred.[10] Unrooted trees can always be generated from rooted ones by simply omitting the root. By contrast, inferring the root of an unrooted tree requires some means of identifying ancestry. This is normally done by including an outgroup in the input data so that the root is necessarily between the outgroup and the rest of the taxa in the tree, or by introducing additional assumptions about the relative rates of evolution on each branch, such as an application of the molecular clock hypothesis.[11]
Bifurcating versus multifurcating
Both rooted and unrooted trees can be either bifurcating or multifurcating. A rooted bifurcating tree has exactly two descendants arising from each interior node (that is, it forms a binary tree), and an unrooted bifurcating tree takes the form of an unrooted binary tree, a free tree with exactly three neighbors at each internal node. In contrast, a rooted multifurcating tree may have more than two children at some nodes and an unrooted multifurcating tree may have more than three neighbors at some nodes.
Labeled versus unlabeled
Both rooted and unrooted trees can be either labeled or unlabeled. A labeled tree has specific values assigned to its leaves, while an unlabeled tree, sometimes called a tree shape, defines a topology only. Some sequence-based trees built from a small genomic locus, such as Phylotree,[12] feature internal nodes labeled with inferred ancestral haplotypes.
Enumerating trees

The number of possible trees for a given number of leaf nodes depends on the specific type of tree, but there are always more labeled than unlabeled trees, more multifurcating than bifurcating trees, and more rooted than unrooted trees. The last distinction is the most biologically relevant; it arises because there are many places on an unrooted tree to put the root. For bifurcating labeled trees, the total number of rooted trees is:
- for , represents the number of leaf nodes.[13]



For bifurcating labeled trees, the total number of unrooted trees is:[13]
- for .


Among labeled bifurcating trees, the number of unrooted trees with leaves is equal to the number of rooted trees with leaves.[4]

The number of rooted trees grows quickly as a function of the number of tips. For 10 tips, there are more than possible bifurcating trees, and the number of multifurcating trees rises faster, with ca. 7 times as many of the latter as of the former.

| Labeled leaves | Binary unrooted trees | Binary rooted trees | Multifurcating rooted trees | All possible rooted trees |
|---|---|---|---|---|
| 1 | 1 | 1 | 0 | 1 |
| 2 | 1 | 1 | 0 | 1 |
| 3 | 1 | 3 | 1 | 4 |
| 4 | 3 | 15 | 11 | 26 |
| 5 | 15 | 105 | 131 | 236 |
| 6 | 105 | 945 | 1,807 | 2,752 |
| 7 | 945 | 10,395 | 28,813 | 39,208 |
| 8 | 10,395 | 135,135 | 524,897 | 660,032 |
| 9 | 135,135 | 2,027,025 | 10,791,887 | 12,818,912 |
| 10 | 2,027,025 | 34,459,425 | 247,678,399 | 282,137,824 |
Special tree types
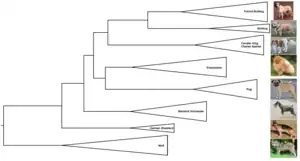
Dendrogram
A dendrogram is a general name for a tree, whether phylogenetic or not, and hence also for the diagrammatic representation of a phylogenetic tree.[14]
Cladogram
A cladogram only represents a branching pattern; i.e., its branch lengths do not represent time or relative amount of character change, and its internal nodes do not represent ancestors.[15]
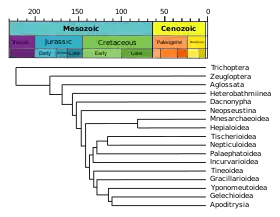
Phylogram
A phylogram is a phylogenetic tree that has branch lengths proportional to the amount of character change.[17]
A chronogram is a phylogenetic tree that explicitly represents time through its branch lengths.[18]
Dahlgrenogram
A Dahlgrenogram is a diagram representing a cross section of a phylogenetic tree.
Phylogenetic network
A phylogenetic network is not strictly speaking a tree, but rather a more general graph, or a directed acyclic graph in the case of rooted networks. They are used to overcome some of the limitations inherent to trees.
Spindle diagram

A spindle diagram, or bubble diagram, is often called a romerogram, after its popularisation by the American palaeontologist Alfred Romer.[19] It represents taxonomic diversity (horizontal width) against geological time (vertical axis) in order to reflect the variation of abundance of various taxa through time. However, a spindle diagram is not an evolutionary tree:[20] the taxonomic spindles obscure the actual relationships of the parent taxon to the daughter taxon[19] and have the disadvantage of involving the paraphyly of the parental group.[21] This type of diagram is no longer used in the form originally proposed.[21]
Coral of life

Darwin[22] also mentioned that the coral may be a more suitable metaphor than the tree. Indeed, phylogenetic corals are useful for portraying past and present life, and they have some advantages over trees (anastomoses allowed, etc.).[21]
Construction
Phylogenetic trees composed with a nontrivial number of input sequences are constructed using computational phylogenetics methods. Distance-matrix methods such as neighbor-joining or UPGMA, which calculate genetic distance from multiple sequence alignments, are simplest to implement, but do not invoke an evolutionary model. Many sequence alignment methods such as ClustalW also create trees by using the simpler algorithms (i.e. those based on distance) of tree construction. Maximum parsimony is another simple method of estimating phylogenetic trees, but implies an implicit model of evolution (i.e. parsimony). More advanced methods use the optimality criterion of maximum likelihood, often within a Bayesian framework, and apply an explicit model of evolution to phylogenetic tree estimation.[4] Identifying the optimal tree using many of these techniques is NP-hard,[4] so heuristic search and optimization methods are used in combination with tree-scoring functions to identify a reasonably good tree that fits the data.
Tree-building methods can be assessed on the basis of several criteria:[23]
- efficiency (how long does it take to compute the answer, how much memory does it need?)
- power (does it make good use of the data, or is information being wasted?)
- consistency (will it converge on the same answer repeatedly, if each time given different data for the same model problem?)
- robustness (does it cope well with violations of the assumptions of the underlying model?)
- falsifiability (does it alert us when it is not good to use, i.e. when assumptions are violated?)
Tree-building techniques have also gained the attention of mathematicians. Trees can also be built using T-theory.[24]
File formats
Trees can be encoded in a number of different formats, all of which must represent the nested structure of a tree. They may or may not encode branch lengths and other features. Standardized formats are critical for distributing and sharing trees without relying on graphics output that is hard to import into existing software. Commonly used formats are
Limitations of phylogenetic analysis
Although phylogenetic trees produced on the basis of sequenced genes or genomic data in different species can provide evolutionary insight, these analyses have important limitations. Most importantly, the trees that they generate are not necessarily correct – they do not necessarily accurately represent the evolutionary history of the included taxa. As with any scientific result, they are subject to falsification by further study (e.g., gathering of additional data, analyzing the existing data with improved methods). The data on which they are based may be noisy;[25] the analysis can be confounded by genetic recombination,[26] horizontal gene transfer,[27] hybridisation between species that were not nearest neighbors on the tree before hybridisation takes place, convergent evolution, and conserved sequences.
Also, there are problems in basing an analysis on a single type of character, such as a single gene or protein or only on morphological analysis, because such trees constructed from another unrelated data source often differ from the first, and therefore great care is needed in inferring phylogenetic relationships among species. This is most true of genetic material that is subject to lateral gene transfer and recombination, where different haplotype blocks can have different histories. In these types of analysis, the output tree of a phylogenetic analysis of a single gene is an estimate of the gene's phylogeny (i.e. a gene tree) and not the phylogeny of the taxa (i.e. species tree) from which these characters were sampled, though ideally, both should be very close. For this reason, serious phylogenetic studies generally use a combination of genes that come from different genomic sources (e.g., from mitochondrial or plastid vs. nuclear genomes),[28] or genes that would be expected to evolve under different selective regimes, so that homoplasy (false homology) would be unlikely to result from natural selection.
When extinct species are included as terminal nodes in an analysis (rather than, for example, to constrain internal nodes), they are considered not to represent direct ancestors of any extant species. Extinct species do not typically contain high-quality DNA.
The range of useful DNA materials has expanded with advances in extraction and sequencing technologies. Development of technologies able to infer sequences from smaller fragments, or from spatial patterns of DNA degradation products, would further expand the range of DNA considered useful.
Phylogenetic trees can also be inferred from a range of other data types, including morphology, the presence or absence of particular types of genes, insertion and deletion events – and any other observation thought to contain an evolutionary signal.
Phylogenetic networks are used when bifurcating trees are not suitable, due to these complications which suggest a more reticulate evolutionary history of the organisms sampled.
See also
- Clade
- Cladistics
- Computational phylogenetics
- Evolutionary biology
- Evolutionary taxonomy
- Generalized tree alignment
- List of phylogenetics software
- List of phylogenetic tree visualization software
- PANDIT, a biological database covering protein domains
- Phylogenetic comparative methods
- Phylogenetic reconciliation
- Taxonomic rank
References
- ↑ Letunic, Ivica; Bork, Peer (1 January 2007). "Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation" (PDF). Bioinformatics. 23 (1): 127–128. doi:10.1093/bioinformatics/btl529. ISSN 1367-4803. PMID 17050570. Archived (PDF) from the original on November 29, 2015. Retrieved 2015-07-21.
- ↑ Ciccarelli, F. D.; Doerks, T.; Von Mering, C.; Creevey, C. J.; Snel, B.; Bork, P. (2006). "Toward automatic reconstruction of a highly resolved tree of life" (PDF). Science. 311 (5765): 1283–1287. Bibcode:2006Sci...311.1283C. CiteSeerX 10.1.1.381.9514. doi:10.1126/science.1123061. PMID 16513982. S2CID 1615592.
- 1 2 Khalafvand, Tyler (2015). Finding Structure in the Phylogeny Search Space. Dalhousie University.
- 1 2 3 4 5 Felsenstein J. (2004). Inferring Phylogenies Sinauer Associates: Sunderland, MA.
- ↑ Kinene, T.; Wainaina, J.; Maina, S.; Boykin, L. (21 April 2016). "Rooting Trees, Methods for". Encyclopedia of Evolutionary Biology: 489–493. doi:10.1016/B978-0-12-800049-6.00215-8. ISBN 9780128004265. PMC 7149615.
- ↑ Bailly, Anatole (1981-01-01). Abrégé du dictionnaire grec français. Paris: Hachette. ISBN 978-2010035289. OCLC 461974285.
- ↑ Bailly, Anatole. "Greek-french dictionary online". www.tabularium.be. Archived from the original on April 21, 2014. Retrieved March 2, 2018.
- ↑ Dang, Cuong Cao; Minh, Bui Quang; McShea, Hanon; Masel, Joanna; James, Jennifer Eleanor; Vinh, Le Sy; Lanfear, Robert (9 February 2022). "nQMaker: Estimating Time Nonreversible Amino Acid Substitution Models". Systematic Biology. 71 (5): 1110–1123. doi:10.1093/sysbio/syac007. PMC 9366462. PMID 35139203.
- ↑ Hodge T, Cope M (1 October 2000). "A myosin family tree". J Cell Sci. 113 (19): 3353–4. doi:10.1242/jcs.113.19.3353. PMID 10984423. Archived from the original on 30 September 2007.
- ↑ ""Tree" Facts: Rooted versus Unrooted Trees". Archived from the original on 2014-04-14. Retrieved 2014-05-26.
- ↑ Maher BA (2002). "Uprooting the Tree of Life". The Scientist. 16 (2): 90–95. Bibcode:2000SciAm.282b..90D. doi:10.1038/scientificamerican0200-90. PMID 10710791. Archived from the original on 2003-10-02.
- ↑ van Oven, Mannis; Kayser, Manfred (2009). "Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation". Human Mutation. 30 (2): E386–E394. doi:10.1002/humu.20921. PMID 18853457. S2CID 27566749.
- 1 2 3 Felsenstein, Joseph (1978-03-01). "The Number of Evolutionary Trees". Systematic Biology. 27 (1): 27–33. doi:10.2307/2412810. ISSN 1063-5157. JSTOR 2412810.
- ↑ Fox, Emily. "The dendrogram". coursea. Archived from the original on 28 September 2017. Retrieved 28 September 2017.
- ↑ Mayr, Ernst (1974)"Cladistic analysis or cladistic classification?". Journal of Zoological Systematics and Evolutionary Research. 12: 94–128. doi:10.1111/j.1439-0469.1974.tb00160.x.
- ↑ Labandeira, C. C.; Dilcher, D. L.; Davis, D. R.; Wagner, D. L. (1994-12-06). "Ninety-seven million years of angiosperm-insect association: paleobiological insights into the meaning of coevolution". Proceedings of the National Academy of Sciences. 91 (25): 12278–12282. Bibcode:1994PNAS...9112278L. doi:10.1073/pnas.91.25.12278. ISSN 0027-8424. PMC 45420. PMID 11607501.
- ↑ Soares, Antonio; Râbelo, Ricardo; Delbem, Alexandre (2017). "Optimization based on phylogram analysis". Expert Systems with Applications. 78: 32–50. doi:10.1016/j.eswa.2017.02.012. ISSN 0957-4174.
- ↑ Santamaria, R.; Theron, R. (2009-05-26). "Treevolution: visual analysis of phylogenetic trees". Bioinformatics. 25 (15): 1970–1971. doi:10.1093/bioinformatics/btp333. PMID 19470585.
- 1 2 "Evolutionary systematics: Spindle Diagrams". Palaeos.com. 2014-11-10. Retrieved 2019-11-07.
- ↑ "Trees, Bubbles, and Hooves". A Three-Pound Monkey Brain — Biology, programming, linguistics, phylogeny, systematics …. 2007-11-21. Retrieved 2019-11-07.
- 1 2 3 Podani, János (2019-06-01). "The Coral of Life". Evolutionary Biology. 46 (2): 123–144. doi:10.1007/s11692-019-09474-w. hdl:10831/46308. ISSN 1934-2845.
- ↑ Darwin, Charles (1837). Notebook B. p. 25.
- ↑ Penny, D.; Hendy, M. D.; Steel, M. A. (1992). "Progress with methods for constructing evolutionary trees". Trends in Ecology and Evolution. 7 (3): 73–79. doi:10.1016/0169-5347(92)90244-6. PMID 21235960.
- ↑ A. Dress, K. T. Huber, and V. Moulton. 2001. Metric Spaces in Pure and Applied Mathematics. Documenta Mathematica LSU 2001: 121-139
- ↑ Townsend JP, Su Z, Tekle Y (2012). "Phylogenetic Signal and Noise: Predicting the Power of a Data Set to Resolve Phylogeny". Genetics. 61 (5): 835–849. doi:10.1093/sysbio/sys036. PMID 22389443.
- ↑ Arenas M, Posada D (2010). "The effect of recombination on the reconstruction of ancestral sequences". Genetics. 184 (4): 1133–1139. doi:10.1534/genetics.109.113423. PMC 2865913. PMID 20124027.
- ↑ Woese C (2002). "On the evolution of cells". Proc Natl Acad Sci USA. 99 (13): 8742–7. Bibcode:2002PNAS...99.8742W. doi:10.1073/pnas.132266999. PMC 124369. PMID 12077305.
- ↑ Parhi, J.; Tripathy, P.S.; Priyadarshi, H.; Mandal, S.C.; Pandey, P.K. (2019). "Diagnosis of mitogenome for robust phylogeny: A case of Cypriniformes fish group". Gene. 713: 143967. doi:10.1016/j.gene.2019.143967. PMID 31279710. S2CID 195828782.
Further reading
- Schuh, R. T. and A. V. Z. Brower. 2009. Biological Systematics: principles and applications (2nd edn.) ISBN 978-0-8014-4799-0
- Manuel Lima, The Book of Trees: Visualizing Branches of Knowledge, 2014, Princeton Architectural Press, New York.
- MEGA, a free software to draw phylogenetic trees.
- Gontier, N. 2011. "Depicting the Tree of Life: the Philosophical and Historical Roots of Evolutionary Tree Diagrams." Evolution, Education, Outreach 4: 515–538.
External links
Images
General
- An overview of different methods of tree visualization is available at Page, R. D. M. (2011). "Space, time, form: Viewing the Tree of Life". Trends in Ecology & Evolution. 27 (2): 113–120. doi:10.1016/j.tree.2011.12.002. PMID 22209094.
- OneZoom: Tree of Life – all living species as intuitive and zoomable fractal explorer (responsive design)
- Discover Life An interactive tree based on the U.S. National Science Foundation's Assembling the Tree of Life Project
- PhyloCode
- A Multiple Alignment of 139 Myosin Sequences and a Phylogenetic Tree
- Tree of Life Web Project
- Phylogenetic inferring on the T-REX server
- NCBI's Taxonomy Database
- ETE: A Python Environment for Tree Exploration This is a programming library to analyze, manipulate and visualize phylogenetic trees. Ref.
- A daily-updated tree of (sequenced) life Fang, H.; Oates, M. E.; Pethica, R. B.; Greenwood, J. M.; Sardar, A. J.; Rackham, O. J. L.; Donoghue, P. C. J.; Stamatakis, A.; De Lima Morais, D. A.; Gough, J. (2013). "A daily-updated tree of (sequenced) life as a reference for genome research". Scientific Reports. 3: 2015. Bibcode:2013NatSR...3E2015F. doi:10.1038/srep02015. PMC 6504836. PMID 23778980.
| Relevant fields | ||
|---|---|---|
| Basic concepts | ||
| Inference methods | ||
| Current topics | ||
| Group traits | ||
| Group types | ||
| Nomenclature | ||
| ||