

Parylene is the common name of a polymer whose backbone consists of para-benzenediyl rings –C
6H
4– connected by 1,2-ethanediyl bridges –CH
2–CH
2–. It can be obtained by polymerization of para-xylylene H
2C=C
6H
4=CH
2.
The name is also used for several polymers with the same backbone, where some hydrogen atoms are replaced by other functional groups. Some of these variants are designated in commerce by letter-number codes such as "parylene C" and "parylene AF-4". Some of these names are registered trademarks in some countries.
Coatings of parylene are often applied to electronic circuits and other equipment as electrical insulation, moisture barriers, or protection against corrosion and chemical attack (conformal coating). They are also used to reduce friction, and in medicine to prevent adverse reactions to implanted devices. These coatings are typically applied by chemical vapor deposition in an atmosphere of the monomer para-xylylene.
Parylene is considered a "green" polymer because its polymerization needs no initiator or other chemicals to terminate the chain; and the coatings can be applied at or near room temperature, without any solvent.
History
Parylene was discovered in 1947 by Michael Szwarc as one of the thermal decomposition products of para-xylene H
3C–C
6H
4–CH
3 above 1000 °C. Szwarc identified para-xylylene as the precursor, by observing that reaction with iodine yielded para-xylylene di-iodide as the only product. The reaction yield was only a few percent.[1][2]
A more efficient route was found in 1965 by William F. Gorham at Union Carbide. He deposited parylene films by the thermal decomposition of [2.2] paracyclophane at temperatures exceeding 550 °C and in vacuum below 1 Torr. This process did not require a solvent and resulted in chemically resistant films free from pinholes. Union Carbide commercialized a parylene coating system in 1965.[1][2]
Union Carbide went on to undertake research into the synthesis of numerous parylene precursors, including parylene AF-4, throughout the 1960s into the early 1970s. Union Carbide purchased NovaTran (a parylene coater) in 1984 and combined it with other electronic chemical coating businesses to form the Specialty Coating Systems division. The division was sold to Cookson Electronics in 1994.[3]
There are parylene coating service companies located around the world, but there is limited commercial availability of parylene. The [2.2]paracyclophane precursors can be purchased for parylene N, C, D, AF-4 and VT-4. Parylene services are provided for N, C, AF-4, VT-4 and E (copolymer of N and E).
Varieties
Parylene N
Parylene N is the un-substituted polymer obtained by polymerization of the para-xylylene intermediate.
Chlorinated parylenes
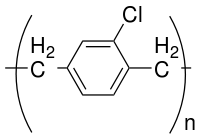
Derivatives of parylene can be obtained by replacing hydrogen atoms on the phenyl ring or the aliphatic bridge by other functional groups. The most common of these variants is parylene C which has one hydrogen atom in the aryl ring replaced by chlorine. Another common variant is parylene D, with two such substitutions on the ring.
Parylene C is the most used variety, due to its low cost of its precursor and to the balance of its properties as dielectric and moisture barrier properties and ease of deposition. A major disadvantage for many applications is its insolubility in any solvent at room temperature, which prevents removal of the coating when the part has to be re-worked.
Parylene C is also the most commonly used because of its relatively low cost.[4] It can be deposited at room temperature while still possessing a high degree of conformality and uniformity and a moderate deposition rate in a batch process.
Also, the chlorine on the phenyl ring of the parylene C repeat unit is problematic for RoHS compliance, especially for the printed circuit board manufacture. Moreover, some of the dimer precursor is decomposed by breaking of the aryl-chlorine bond during pyrolysis, generating carbonaceous material that contaminates the coating, and hydrogen chloride HCl that may harm vacuum pumps and other equipment. The chlorine atom leaves the phenyl ring in the pyrolysis tube at all temperatures; however, optimizing the pyrolysis temperature will minimize this problem. The free-radical (phenyl radical) generated in this process is not resonance-stabilized and mitigates the deposition of a parylene-like material on the downside of the pyrolysis tube. This material becomes carbonized and generates particles in situ to contaminate clean rooms and create defects on printed-circuit boards that are often called 'stringers and nodules'. Parylene N and E do not have this problem and therefore are preferred for manufacturing and clean room use.
Fluorinated parylenes
Another common halogenated variant is parylene AF-4, with the four hydrogen atoms on the aliphatic chain replaced by fluorine atoms. This variant is also marketed under the trade names of parylene SF (Kisco) and HT parylene (SCS). The –CF
2– unit that comprises the ethylene chain is the same as the repeating unit of PTFE (Teflon), consistent with its superior oxidative and UV stability. Parylene AF-4 has been used to protect outdoor LED displays and lighting from water, salt and pollutants successfully.
Another fluorinated variant is parylene VT-4 (also called parylene F), with fluorine substituted for the four hydrogens on the aryl ring. This variant is marketed by Kisco with the trademark Parylene CF. Because of the aliphatic -CH2- units, it has poor oxidative and UV stability, but still better than N, C, or D.
Alkyl-substituted parylenes
The hydrogen atoms can be replaced also by alkyl groups. Substitution may occur on either the phenyl ring or the ethylene bridge, or both.
Specifically, replacement of one hydrogen on the phenyl ring by a methyl group or an ethyl group yields parylene M and parylene E, respectively.
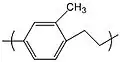 Parylene M
Parylene M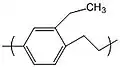 Parylene E
Parylene E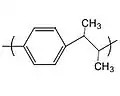 Parylene AM-2
Parylene AM-2
These substitutions increase the intermolecular (chain-to-chain) distance, which makes the polymer more soluble and permeable. For example, compared to parylene C, parylene M was shown to have a lower dielectric constant (2.48 vs. 3.2 at 1 kHz). Parylene E had a lower tensile modulus (175 kpi vs. 460 kpsi), a lower dielectric constant (2.34 vs. 3.05 at 10 kHz), slightly worse moisture barrier properties (4.1 vs. 0.6 g-mil/atom-100in2-24hr), and equivalent dielectric breakdown 5-6 kvol/mil for a 1 mil coating) but better solubility.[5][6] However, the copolymer of parylene N and E has equivalent barrier performance of parylene C.
Replacement of one hydrogen by methyl on each carbon of the ethyl bridge yields parylene AM-2, [–(CH
3)CH–(C
6H
4)–(CH
3)CH–]
n (not to be confused with an amine-substituted variant trademarked by Kisco). The solubility of parylene AM-2 is not as good as parylene E.
Reactive parylenes
While parylene coatings are mostly used to protect an object from water and other chemicals, some applications require a coating that can bind to adhesives or other coated parts, or immobilize various molecules such as dyes, catalysts, or enzymes.
These "reactive" parylene coatings can be obtained with chemically active substituents. Two commercially available products are parylene A, featuring one amine substituent –NH
2 in each unit, and parylene AM, with one methylene amine group –CH
2NH
2 per unit. (Both are trademarks of Kisco.)
Parylene AM is more reactive than the A variant. The amine of the latter, being adjacent to the phenyl ring is in resonance stabilization and therefore less basic. However, parylene A is much easier to synthesize and hence it costs less.

Another reactive variant is parylene X, which features an ethinyl group –C≡CH attached to the phenyl ring in some of the units. This variant, which contains no elements other than hydrogen and carbon, can be cross-linked by heat or with UV light, and can react with copper or silver salts to generate the corresponding metalorganic complexes Cu-acetylide or Ag-acetylide. It can also undergo 'click chemistry', and can be used as an adhesive, allowing parylene-to-parylene bonding without any by-products during processing. Unlike most other variants, parylene X is amorphous (non-crystalline).
Colored parylenes
It is possible to attach a chromophore directly to the [2.2]paracyclophane base molecule to impart color to parylene.
Parylene-like copolymers
Copolymers[7] and nanocomposites (SiO2/parylene C)[8] of parylene have been deposited at near-room temperature previously; and with strongly electron withdrawing comonomers, parylene can be used as an initiator to initiate polymerizations, such as with N-phenyl maleimide. Using the parylene C/SiO2 nanocomposites, parylene C could be used as a sacrificial layer to make nanoporous silica thin films with a porosity of >90%.[9]
Properties
Transparency and crystallinity
Parylene thin films and coatings are transparent; however, they are not amorphous except for the alkylated parylenes. i.e. parylene E. As a result, of the coatings being semi-crystalline, they scatter light. Parylene N and C have a low degree of crystallinity; however, parylene VT-4 and AF-4 are highly crystalline ~60% in their as-deposited condition (hexagonal crystal structure) and therefore are generally not suitable as optical materials.
Parylene C will become more crystalline if heated at elevated temperatures until its melting point at 270 °C.
Parylene N has a monoclinic crystal structure in its as-deposited condition and it does not appreciably become more crystalline until it undergoes a crystallographic phase transformation at ~220 °C to hexagonal, at which point it becomes highly crystalline like the fluorinated parylenes. It can reach 80% crystallinity at anneal temperatures up to 400 °C, after which point it degrades.
Mechanical and chemical
Parylenes are relatively flexible (parylene N 0.5 GPa)[10] except for cross-linked parylene X (1.0 GPa)[11] and they have poor oxidative resistance (~60-100 °C depending on failure criteria) and UV stability,[12] except for parylene AF-4. However, parylene AF-4 is more expensive due to a three-step synthesis of its precursor with low yield and poor deposition efficiency. Their UV stability is so poor that parylene cannot be exposed to regular sunlight without yellowing.
Nearly all the parylenes are insoluble at room temperature except for the alkylated parylenes, one of which is parylene E [6] and the alkylated-ethynyl parylenes.[13] This lack of solubility has made it difficult to re-work printed circuit boards coated with parylene.
Permeability
As a moisture diffusion barrier, the efficacy of halogneated parylene coatings scales non-linearly with their density. Halogen atoms such as F, Cl and Br add much density to the coating and therefore allow the coating to be a better diffusion barrier; however, if parylenes are used as a diffusion barrier against water then the apolar chemistries such as parylene E are much more effective. For moisture barriers the three principal material parameters to be optimized are: coating density, coating polarity (olefin chemistry is best) and a glass-transition temperature above room temperature and ideally above the service limit of the printed-circuit board, device or part. In this regard parylene E is a best choice although it has a low density compared to, for example, parylene C.
Industry Spec[14]
| Properties | Parylene N | Parylene C | Parylene D | Parylene HT/AF4 |
|---|---|---|---|---|
| Melting Point (°C) | 420 | 290 | 380 | >500 |
| Continuous Service Temperature (°C) | 60 | 80 | 100 | 350 |
| Short-Term Service Temperature (°C) | 80 | 100 | 120 | 450 |
| Linear Coefficient of Thermal Expansion at 25°C (ppm) | 69 | 35 | 38 | 36 |
| Thermal Conductivity at 25°C (W/(m·K)) | 0.126 | 0.084 | - | 0.096 |
| Specific Heat at 20°C (J/(g·K)) | 0.837 | 0.712 | - | 1.04 |
| Young's Modulus (psi) | 350,000 | 400,000 | 380,000 | 370,000 |
| Tensile Strength (psi) | 7,000 | 10,000 | 11,000 | 7,500 |
| Yield Strength (psi) | 6,100 | 8,000 | 9,000 | 5,000 |
| Elongation to Break (%) | 250 | 200 | 200 | 200 |
| Yield Elongation (%) | 2.5 | 2.9 | 3.0 | 2.0 |
| Density (g/cm3) | 1.1-1.12 | 1.289 | 1.418 | 1.32 |
| Water Absorption (% after 24 hours) | <0.1 | <0.1 | <0.1 | <0.01 |
| Rockwell Hardness | R85 | R80 | R80 | R122 |
| Static Coefficient of Friction | 0.25 | 0.29 | 0.33 | 0.15 |
| Dynamic Coefficient of Friction | 0.25 | 0.29 | 0.31 | 0.13 |
Coating process
Parylene coatings are generally applied by chemical vapor deposition in an atmosphere of the monomer para-xylylene or a derivative thereof. This method has one very strong benefit, namely it does not generate any byproducts besides the parylene polymer, which would need to be removed from the reaction chamber and could interfere with the polymerization.
Parts to be coated need to be clean in order to ensure good adherence of the film. Since the monomer diffuses, areas that are not to be coated must be hermetically sealed, without gaps, crevices or other openings. The part must be maintained in a relatively narrow window of pressure and temperature.[15]
The process involves three steps: generation of the gaseous monomer, adsorption on the part's surface, and polymerization of adsorbed film.
Polymerization
Polymerization of the adsorbed p-xylylene monomer requires a minimum threshold temperature. For parylene N, its threshold temperature is 40 °C.
The p-xylylene intermediate has two quantum mechanical states, the benzoid state (triplet state) and the quinoid state (singlet state). The triplet state is effectively the initiator and the singlet state is effectively the monomer. The triplet state can be de-activated when in contact with transition metals or metal oxides including Cu/CuOx.[16][17] Many of the parylenes exhibit this selectivity based on quantum mechanical deactivation of the triplet state, including parylene X.
Polymerization may proceed by a variety of routes that differ in the transient termination of the growing chains, such as a radical group –CH•
2 or a negative anion group CH−
2:

Physisorption
The monomer polymerizes only after it is physically adsorbed (physisorbed) on the part's surface. This process has inverse Arrhenius kinetics, meaning that it is stronger at lower temperatures than higher temperatures. There is critical threshold temperature above which there is practically no physisorption, and hence no deposition. The closer the deposition temperature is to the threshold temperature the weaker the physisorption. Parylene C has a higher threshold temperature, 90 °C, and therefore has a much higher deposition rate, greater than 1 nm/s, while still yielding fairly uniform coatings.[4] In contrast, the threshold temperature of parylene AF-4 is very close to room temperature (30–35 °C), as a result, its deposition efficiency is poor.[18]
An important property of the monomer is the so-called 'sticking coefficient', that expresses the degree to which it adsorbs on the polymer. A lower coefficient results more uniform deposition thickness and a more conformal coating.
Another relevant property for the deposition process is polarizability, which determines how strongly the monomer interacts with the surface. Deposition of halogenated parylenes strongly correlates with molecular weight of the monomer. The fluorinated variants are an exception: the polarizability of parylene AF-4 is low, resulting in inefficient deposition.
Monomer generation
From the cyclic dimer

The p-xylylene monomer is normally generated during the coating process by evaporating the cyclic dimer [2.2]para-cyclophane at a relatively low temperature, then decomposing the vapor at 450–700 °C and pressure 0.01–1.0 torr. This method (Gorham Process) yields 100% monomer with no by-products or decomposition of the monomer. [19][20][21]
The dimer can be synthesized from p-xylene involving several steps involving bromination, amination and Hofmann elimination.[22]
The same method can be used to deposit substituted parylenes. For example, parylene C can be obtained from the dimeric precursor dichloro[2.2]para-cyclophane, except that the temperature must be carefully controlled since the chlorine-aryl bond breaks at 680 °C.
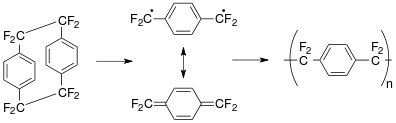
The standard Gorham process[5] is shown above for parylene AF-4. The octafluoro[2.2]para-cyclophane precursor dimer can be sublimed below <100 °C and cracked at 700-750 °C, higher than the temperature (680 °C) used to crack the unsubstituted cyclophane since the -CF2-CF2- bond is stronger than the -CH2-CH2- bond. This resonance-stabilized intermediate is transported to a room temperature deposition chamber where polymerization occurs under low pressure (1–100 mTorr) conditions.[18]
From substituted p-xylenes
Another route to generation of the monomer is to use a para-xylene precursor with a suitable substituent on each methyl groups, whose elimination generates para-xylylene.
Selection of a leaving group may consider its toxicity (which excludes sulfur and amine-based reactions), how easily it leaves the precursor, and possible interference with the polymerization. The leaving group can either be trapped before the deposition chamber, or it can be highly volatile so that it does not condense in the latter.[23]
For example, the precursor α,α'-dibromo-α,α,α',α'-tetrafluoro-para-xylene (CF
2Br)
2(C
6H
4) yields parylene AF-4 with elimination of bromine.[24]

The advantage to this process is the low cost of synthesis for the precursor. The precursor is also a liquid and can be delivered by standard methods developed in the semiconductor industry, such as with a vaporizer, vaporizer with a bubbler, or a mass-flow controller. Originally the precursor was just thermally cracked,[25] but suitable catalysts lower the pyrolysis temperature, resulting in less char residue and a better coating.[26][27] By either method an atomic bromine free-radical is given off from each methyl end, which can be converted to hydrogen bromide HBr and removed from monomer flow. Special precautions are needed since bromine and HBr are toxic and corrosive towards most metals and metal alloys, and bromine can damage viton O-rings.

A similar synthesis for parylene N uses the precursor α,α'-dimethoxy-p-xylene.[28] The methoxy group H
3CO– is the leaving group; while it condenses in the deposition chamber, it does not interfere with the deposition of the polymer.[23] This precursor is much less expensive than [2.2]para-cyclophane. Moreover, being a liquid just above room temperature, this precursor can delivered reliably using a mass-flow controller; whereas the generation and delivery of the gaseous monomer of the Gorham process are difficult to measure and control.[29]

The same chemistry can generate parylene AM-2 can be generated from the precursor α,α'-dimethyl-α,α'-dimethoxy-p-xylene.
Another example of this approach is the synthesis of parylene AF-4 from α,α'-diphenoxy-α,α,α',α'-tetrafluoro-para-xylene. In this case, the leaving group is phenoxy CH
5O–, which can be condensed before the deposition chamber.[30]
Characteristics and advantages
Parylenes may confer several desirable qualities to the coated parts. Among other properties, they are
- Hydrophobic, chemically resistant, and mostly impermeable to gases (including water vapor) and inorganic and organic liquids (including strong acids and bases).
- Good electrical insulator with a low dielectric constant (average in-plane and out-of-plane: 2.67 parylene N and 2.5 parylene AF-4, SF, HT)[31]
- Stable and accepted in biological tissues, having been approved by the US FDA for various medical applications.
- Dense and pinhole free, for thickness above 1.4 nm[32]
- Homogeneous and uniformly thick, even within cavities.
- Stable to oxidation up to 350 °C (AF-4, SF, HT)
- Low coefficient of friction (AF-4, HT, SF)
Since the coating process takes place at ambient temperature in a mild vacuum, it can be applied even to temperature-sensitive objects such as dry biological specimens. The low temperature also results in low intrinsic stress in the thin film. Moreover, the only gas in the deposition chamber is the monomer, without any solvents, catalysts, or byproducts that could attack the object.
Parylene AF-4 and VT-4 are both fluorinated and as a result very expensive compared to parylene N and C, which has severely limited their commercial use, except for niche applications.
Applications
Parylene C and to a lesser extent AF-4, SF, HT (all the same polymer) are used for coating printed circuit boards (PCBs) and medical devices. There are numerous other applications as parylene is an excellent moisture barrier. It is the most bio-accepted coating for stents, defibrillators, pacemakers and other devices permanently implanted into the body.[33]
Molecular layers
The classic molecular layer chemistries are self-assembled monolayers (SAMs). SAMs are long-chain alkyl chains, which interact with surfaces based on sulfur-metal interaction (alkylthiolates)[34] or a sol-gel type reaction with a hydroxylated oxide surface (trichlorosilyl alkyls or trialkoxy alkyls).[35] However, unless the gold or oxide surface is carefully treated and the alkyl chain is long, these SAMs form disordered monolayers, which do not pack well.[36][37] This lack of packing causes issues in, for example, stiction in MEMS devices.[38]
The observation that parylenes could form ordered molecular layers (MLs) came with contact angle measurements, where MLs thicker than 10 Å had an equilibrium contact angle of 80 degrees (same as bulk parylene N) but those thinner had a reduced contact angle.[32] This was also confirmed with electrical measurements (bias-temperature stress measurements) using metal-insulator-semiconductor capacitors (MISCAPs).[39] In short, parylene N and AF-4 (those parylenes with no functional groups) are pin-hole free at ~14 Å. This results because the parylene repeat units possess a phenyl ring and due to the high electronic polarizability of the phenyl ring adjacent repeat units order themselves in the XY-plane. As a result of this interaction parylene MLs are surface independent, except for transition metals, which de-activate the triplet (benzoid) state and therefore the parylenes cannot be initiated. This finding of parylenes as molecular layers is very powerful for industrial applications because of the robustness of the process and that the MLs are deposited at room temperature. In this way parylenes can be used as diffusion barriers and for reducing the polarizability of surface (de-activation of oxide surfaces). Combining the properties of the reactive parylenes with the observation that they can form dense pin-hole-free molecular layers, parylene X has been utilized as a genome sequencing interface layer.
One caveat with the molecular layer parylenes, namely they are deposited as oligomers and not high polymer.[32] As a result, a vacuum anneal is needed to convert the oligomers to high polymer. For parylene N that temperature is 250 °C, whereas it is 300 °C for payrlene AF-4.
Typical applications
Parylene films have been used in various applications, including[1]
- Hydrophobic coating (moisture barriers, e.g., for biomedical hoses)
- Barrier layers (e.g., for filter, diaphragms, valves)
- Microwave electronics (e.g., protection of PTFE dielectric substrates from oil contamination)
- Implantable medical devices
- Sensors in rough environment (e.g., automotive fuel/air sensors)
- Electronics for space travel and defense
- Corrosion protection for metallic surfaces
- Reinforcement of micro-structures
- Protection of plastic, rubber, etc., from harmful environmental conditions
- Reduction of friction, e.g., for guiding catheters, acupuncture needles and microelectromechanical systems.
See also
References
- 1 2 3 Jeffrey B. Fortin; Toh-Ming Lu (2003). Chemical vapor deposition polymerization: the growth and properties of parylene thin films. Springer. pp. 4–7. ISBN 978-1-4020-7688-6.
- 1 2 Mattox, D. M. The foundations of vacuum coating technology Archived 2009-10-07 at the Wayback Machine, Springer, 2003 ISBN 978-3-540-20410-7 Google books
- ↑ SCS Coatings History Archived 2012-01-12 at the Wayback Machine. Scscoatings.com. Retrieved on 2012-06-04.
- 1 2 "Parylene: The Truly Conformal Thin Film Coating" (PDF). Plasma Ruggedized Solutions. PRS.
- 1 2 W. F. Gorham (1966). "A New, General Synthetic Method for the Preparation of Linear Poly-p-xylylenes". Journal of Polymer Science Part A-1: Polymer Chemistry. 4 (12): 3027–3039. Bibcode:1966JPoSA...4.3027G. doi:10.1002/pol.1966.150041209.
- 1 2 J. J. Senkevich; C. J. Mitchell; A. Vijayaraghavan; E. V. Barnat; J. F. McDonald; T.-M. Lu (2002). "The Unique Structure/Properties of Chemical Vapor Deposited Parylene E". Journal of Vacuum Science and Technology A. 20 (4): 1445–9. Bibcode:2002JVSTA..20.1445S. doi:10.1116/1.1487870.
- ↑ J. F. Gaynor; J. J. Senkevich; S. B. Desu (1996). "A New Method for Fabricating High Performance Polymeric Thin Films by Chemical Vapor Polymerization". J. Mater. Res. 11 (7): 1842–50. Bibcode:1996JMatR..11.1842G. doi:10.1557/JMR.1996.0233.
- ↑ J.J. Senkevich; S. B. Desu (1999). "Near-Room-Temperature Thermal Chemical Vapor Deposition of Poly(chloro-p-xylylene)/SiO2 Nanocomposites". Chemistry of Materials. 11 (7): 1814–21. doi:10.1021/cm990042q.
- ↑ J. J. Senkevich (1999). "CVD of NanoPorous Silica". Chemical Vapor Deposition. 5 (6): 257–60. doi:10.1002/(SICI)1521-3862(199912)5:6<257::AID-CVDE257>3.0.CO;2-J.
- ↑ C. Chiang, A. S. Mack, C. Pan, Y.-L. Ling, D. B. Fraser Mat. Res. Soc. Symp. Proc. vol. 381, 123 (1995).
- ↑ J. J. Senkevich; B. W. Woods; J. J. McMahon; P.-I Wang (2007). "Thermomechanical Properties of Parylene X, A Room-Temperature Chemical Vapor Depositable Crosslinkable Polymer". Chemical Vapor Deposition. 13 (1): 55–59. doi:10.1002/cvde.200606541.
- ↑ J.B. Fortin & T.-M. Lu (2001). "Ultraviolet radiation induced degradation of poly-para-xylylene (parylene) thin films". Thin Solid Films. 397 (1–2): 223–228. Bibcode:2001TSF...397..223F. doi:10.1016/S0040-6090(01)01355-4.
- ↑ Senkevich, Jay J. (2014). "Tert-Butylethynyl-parylene and Phenylethynyl-parylene". Chemical Vapor Deposition. 20 (1–2–3): 39–43. doi:10.1002/cvde.201307071.
- ↑ "Specialty Coating Systems - Parylene Properties" (PDF).
- ↑ Horn, Sean "The Parylene Deposition Process: Pre-Deposition" https://www.paryleneconformalcoating.com/#TheParyleneDepositionProcess
- ↑ K. M. Vaeth & K. F. Jensen (1999). "Selective growth of poly(p-phenylene vinylene) prepared by chemical vapor deposition". Advanced Materials. 11 (10): 814–820. doi:10.1002/(SICI)1521-4095(199907)11:10<814::AID-ADMA814>3.0.CO;2-Z.
- ↑ J.J. Senkevich; C.J. Wiegand; G.-R. Yang; T.-M. Lu (2004). "Selective Deposition of Ultra-thin Poly(p-xylylene) Films on Dielectrics versus Copper Surfaces". Chemical Vapor Deposition. 10 (5): 247–9. doi:10.1002/cvde.200304179.
- 1 2 "GlobalTop Technology | Taiwan | Aluminum Nitride Powder".
- ↑ J. B. Fortin & T.-M. Lu (2000). "Mass spectrometry study during the vapor deposition of poly-para-xylylene thin films". Journal of Vacuum Science and Technology A. 18 (5): 2459. Bibcode:2000JVSTA..18.2459F. doi:10.1116/1.1289773.
- ↑ H. J. Reich; D. J. Cram (1969). "Macro rings. XXXVI. Ring expansion, racemization, and isomer interconversions in the [2.2]paracyclophane system through a diradical intermediate". Journal of the American Chemical Society. 91 (13): 3517–3526. doi:10.1021/ja01041a016.
- ↑ P. Kramer; A. K. Sharma; E. E. Hennecke; H. Yasuda (2003). "Polymerization of para-xylylene derivatives (parylene polymerization). I. Deposition kinetics for parylene N and parylene C". Journal of Polymer Science: Polymer Chemistry Edition. 22 (2): 475–491. Bibcode:1984JPoSA..22..475K. doi:10.1002/pol.1984.170220218.
- ↑ H. E. Winberg and F. S. Fawcett (1973). "Tricyclo[8.2.2.24,7]hexadeca-4,6,10,12,13,15-hexaene". Organic Syntheses.
- 1 2 J.J. Senkevich (2011): "CVD of Poly(α,α'-dimethyl-p-xylylene and Poly(α,α,α',α'-tetramethyl-p-xylylene)-co-poly(p-xylylene) from Alkoxide Precursors I: Optical Properties and Thermal Stability". Chemical Vapor Deposition, volume 17, pages 235-240.
- ↑ P. K. Wu; G. -R. Yang; L. You; D. Mathur; A. Cocoziello; C. -I. Lang; J. A. Moore; T. -M. Lu; H. Bakru (1997). "Deposition of High Purity Parylene- F Using Low Pressure Low Temperature Chemical Vapor Deposition". Journal of Electronic Materials. 26 (8): 949–953. Bibcode:1997JEMat..26..949W. doi:10.1007/s11664-997-0280-8. S2CID 94987047.
- ↑ Pebalk, A. V.; Kardash, I. E.; Kozlova, N. V.; Zaitseva, E. L.; Kozlov, Yu. A.; Pravednikov, A. N. (1980). Vysokomolekulyarnye Soedineniya, Seriya A. 22 (5): 972–6.
{{cite journal}}: Missing or empty|title=(help) - ↑ Lee, Chung J.; Wang, Hui; Foggiato, Giovanni Antonio, U.S. Patent 6,140,456, Issue date: October 31, 2000.
- ↑ Lee, Chung J., U.S. Patent 6,703,462, Issue date: March 9, 2004.
- ↑ Senkevich, Jay J. (2011). "Non-Halogen Liquid Precursor Route to Parylene". Chemical Vapor Deposition. 17 (4–6): 76–79. doi:10.1002/cvde.201104304.
- ↑ D.M. Dobkin, S. Mokhtari, M. Schmidt, A. Pant, L. Robinson, Mechanisms of Deposition of SiO2 from TEOS and Related Organosilicon Compounds and Ozone" J. Electrochem. Soc. 142(7), 2332-40 (1995).
- ↑ Senkevich, Jay J. (2013). "Parylene AF-4 via the Trapping of a Phenoxy Leaving Group". Chemical Vapor Deposition. 19 (10–11–12): 327–331. doi:10.1002/cvde.201304321.
- ↑ J. J. Senkevich; S. B. Desu (1999). "Compositional studies of near-room temperature thermal CVD of poly(chloro-p-xylylene)/SiO2 nanocomposites". Chemistry of Materials. 11 (5): 1814. Bibcode:2000ApPhA..70..541S. doi:10.1007/s003390051076. S2CID 96072554.
- 1 2 3 J. J. Senkevich & P.-I. Wang (2009). "Molecular Layer Chemistry via Parylenes". Chemical Vapor Deposition. 15 (4–6): 91–94. doi:10.1002/cvde.200804266.
- ↑ James A. Schwarz; Cristian I. Contescu; Karol Putyera (2004). Dekker encyclopédia of nanoscience and nanotechnology, Volume 1. CRC Press. p. 263. ISBN 978-0-8247-5047-3.
- ↑ Laibinis, Paul E.; Whitesides, George M.; Allara, David L.; Tao, Yu Tai; Parikh, Atul N.; Nuzzo, Ralph G. (1991). "Comparison of the structures and wetting properties of self-assembled monolayers of n-alkanethiols on the coinage metal surfaces, copper, silver, and gold". Journal of the American Chemical Society. 113 (19): 7152. doi:10.1021/ja00019a011.
- ↑ Wasserman, Stephen R.; Tao, Yu Tai; Whitesides, George M. (1989). "Structure and reactivity of alkylsiloxane monolayers formed by reaction of alkyltrichlorosilanes on silicon substrates". Langmuir. 5 (4): 1074. doi:10.1021/la00088a035.
- ↑ Fadeev, Alexander Y.; McCarthy, Thomas J. (2000). "Self-Assembly is Not the Only Reaction Possible between Alkyltrichlorosilanes and Surfaces: Monomolecular and Oligomeric Covalently Attached Layers of Dichloro- and Trichloroalkylsilanes on Silicon". Langmuir. 16 (18): 7268. doi:10.1021/la000471z.
- ↑ Senkevich, Jay J.; Mitchell, Christopher J.; Yang, G.-R.; Lu, T.-M. (2002). "Surface Chemistry of Mercaptan and Growth of Pyridine Short-Chain Alkoxy Silane Molecular Layers". Langmuir. 18 (5): 1587. doi:10.1021/la010970f.
- ↑ Z. Yapu (2003). "Stiction and anti-stiction in MEMS and NEMS". Acta Mechanica Sinica. 19 (1): 1. Bibcode:2003AcMSn..19....1Z. doi:10.1007/BF02487448. S2CID 110540114.
- ↑ Senkevich, Jay J.; Wang, Pei-I.; Wiegand, Chris J.; Lu, T.-M. (2004). "Bias-Temperature Stability of Ultra Thin Parylene Capped PETEOS Dielectrics: Influence of Surface Oxygen on Copper Ion Diffusion". Applied Physics Letters. 84 (14): 2617. Bibcode:2004ApPhL..84.2617S. doi:10.1063/1.1691488.