



| Cyclin-dependent kinase inhibitor 2a p19Arf N-terminus | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 solution structure of the n-terminal 37 amino acids of the mouse arf tumor suppressor protein | |||||||||
| Identifiers | |||||||||
| Symbol | P19Arf_N | ||||||||
| Pfam | PF07392 | ||||||||
| InterPro | IPR010868 | ||||||||
| SCOP2 | 1hn3 / SCOPe / SUPFAM | ||||||||
| |||||||||
p16 (also known as p16INK4a, cyclin-dependent kinase inhibitor 2A, CDKN2A, multiple tumor suppressor 1 and numerous other synonyms), is a protein that slows cell division by slowing the progression of the cell cycle from the G1 phase to the S phase, thereby acting as a tumor suppressor. It is encoded by the CDKN2A gene. A deletion (the omission of a part of the DNA sequence during replication) in this gene can result in insufficient or non-functional p16, accelerating the cell cycle and resulting in many types of cancer.[5][6][7]
p16 can be used as a biomarker to improve the histological diagnostic accuracy of grade 3 cervical intraepithelial neoplasia (CIN). p16 is also implicated in the prevention of melanoma, oropharyngeal squamous cell carcinoma, cervical cancer, vulvar cancer and esophageal cancer.
p16 was discovered in 1993. It is a protein with 148 amino acids and a molecular weight of 16 kDa that comprises four ankyrin repeats.[8] The name of p16 is derived from its molecular weight, and the alternative name p16INK4a refers to its role in inhibiting cyclin-dependent kinase CDK4.[8]
Nomenclature
p16 is also known as:
- p16INK4A
- p16Ink4
- Cyclin-dependent kinase inhibitor 2A (CDKN2A)
- CDKN2
- CDK 4 Inhibitor
- Multiple Tumor Suppressor 1 (MTS1)
- TP16
- ARF
- MLM
- P14
Gene
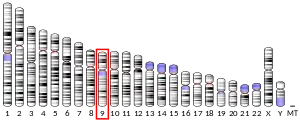
In humans, p16 is encoded by the CDKN2A gene, located on chromosome 9 (9p21.3). This gene generates several transcript variants that differ in their first exons. At least three alternatively spliced variants encoding distinct proteins have been reported, two of which encode structurally related isoforms known to function as inhibitors of CDK4. The remaining transcript includes an alternate exon 1 located 20 kb upstream of the remainder of the gene; this transcript contains an alternate open reading frame (ARF) that specifies a protein that is structurally unrelated to the products of the other variants.[9] The ARF product functions as a stabilizer of the tumor suppressor protein p53, as it can interact with and sequester MDM2, a protein responsible for the degradation of p53.[10][11] In spite of their structural and functional differences, the CDK inhibitor isoforms and the ARF product encoded by this gene, through the regulatory roles of CDK4 and p53 in cell cycle G1 progression, share a common functionality in controlling the G1 phase of the cell cycle. This gene is frequently mutated or deleted in a wide variety of tumors and is known to be an important tumor suppressor gene.[5]
When organisms age, the expression of p16 increases to reduce the proliferation of stem cells.[12] This reduction in the division and production of stem cells protects against cancer while increasing the risks associated with cellular senescence.
Function
p16 is an inhibitor of cyclin-dependent kinases (CDK). It slows down the cell cycle by prohibiting progression from G1 phase to S phase. Otherwise, CDK4/6 binds cyclin D and forms an active protein complex that phosphorylates retinoblastoma protein (pRB). Once phosphorylated, pRB dissociates from the transcription factor E2F1. This liberates E2F1 from its bound state in the cytoplasm and allows it to enter the nucleus. Once in the nucleus, E2F1 promotes the transcription of target genes that are essential for transition from G1 to S phase.[13][14]
This pathway connects the processes of tumor oncogenesis and senescence, fixing them on opposite ends of a spectrum. On one end, p16 hypermethylation, mutation, or deletion leads to downregulation of the gene and can lead to cancer through the dysregulation of cell cycle progression. Conversely, activation of p16 through reactive oxygen species, DNA damage, or senescence leads to the buildup of p16 in tissues and is implicated in the aging of cells.[13]
Regulation
Regulation of p16 is complex and involves the interaction of several transcription factors, as well as several proteins involved in epigenetic modification through methylation and repression of the promoter region.[13]
PRC1 and PRC2 are two protein complexes that modify the expression of p16 through the interaction of various transcription factors that execute methylation patterns that can repress transcription of p16. These pathways are activated in the cellular response to reduce senescence.[15][16]
Clinical significance
Role in carcinogenesis
Mutations resulting in deletion or reduction of function of the CDKN2A gene are associated with increased risk of a wide range of cancers, and alterations of the gene are frequently seen in cancer cell lines.[17][18] Examples include:
Pancreatic adenocarcinoma is often associated with mutations in the CDKN2A gene.[19][20][21]
Carriers of germline mutations in CDKN2A have, besides their high risks of melanoma, also increased risks of pancreatic, lung, laryngeal and oropharyngeal cancers. Tobacco smoking increases the carriers’ susceptibility for such non-melanoma cancers.[22]
Homozygous deletions of p16 are frequently found in esophageal cancer and gastric cancer cell lines.[23]
Germline mutations in CDKN2A are associated with an increased susceptibility to develop skin cancer.[24]
Hypermethylation of tumor suppressor genes has been implicated in various cancers. In 2013, a meta-analysis revealed an increased frequency of DNA methylation of the p16 gene in esophageal cancer. As the degree of tumor differentiation increased, so did the frequency of p16 DNA methylation.
Tissue samples of primary oral squamous cell carcinoma (OSCC) often display hypermethylation in the promoter regions of p16. Cancer cells show a significant increase in the accumulation of methylation in CpG islands in the promoter region of p16. This epigenetic change leads to loss of the tumor suppressor gene function through two possible mechanisms: first, methylation can physically inhibit the transcription of the gene, and second, methylation can lead to the recruitment of transcription factors that repress transcription. Both mechanisms cause the same end result: downregulation of gene expression that leads to decreased levels of the p16 protein. It has been suggested that this process is responsible for the development of various forms of cancer serving as an alternative process to gene deletion or mutation.[25][26][27][28][29][30]
p16 positivity has been shown to be favorably prognostic in oropharyngeal squamous cell carcinoma.[31] In a retrospective trial analysis of patients with Stage III and IV oropharyngeal cancer, HPV status was assessed and it was found that the 3-year rates of overall survival were 82.4% (95% CI, 77.2 to 87.6) in the HPV-positive subgroup and 57.1% (95% CI, 48.1 to 66.1) in the HPV-negative subgroup, and the 3-year rates of progression-free survival were 73.7% (95% CI, 67.7 to 79.8) and 43.4% (95% CI, 34.4 to 52.4), respectively. p16 status is so prognostic that the AJCC staging system has been revised to include p16 status in oropharyngeal squamous cell cancer group staging.[32] However, some people can have elevated levels of p16 but test negative for HPV and vice versa. This is known as discordant cancer. The 5-year survival for people who test positive for HPV and p16 is 81%, for discordant cancer it is 53 – 55%, and 40% for those who test negative for p16 and HPV.[33][34]
Clinical use
Biomarker for cancer types
Expression of p16 is used as a prognostic biomarker for certain types of cancer. The reason for this is different types of cancer can have different effects on p16 expression: cancers that overexpress p16 are usually caused by the human papillomavirus (HPV), whereas cancers in which p16 is downregulated will usually have other causes. For patients with oropharyngeal squamous cell carcinoma, using immunohistochemistry to detect the presence of the p16 biomarker has been shown to be the strongest indicator of disease course. Presence of the biomarker is associated with a more favorable prognosis as measured by cancer-specific survival (CSS), recurrence-free survival (RFS), locoregional control (LRC), as well as other measurements. The appearance of hypermethylation of p16 is also being evaluated as a potential prognostic biomarker for prostate cancer.[35][36][37]
p16 FISH
p16 deletion detected by FISH in surface epithelial mesothelial proliferations is predictive of underlying invasive mesothelioma.[38]
p16 immunochemistry

As consensus grows regarding the strength of p16 as a biomarker for detecting and determining prognoses of cancer, p16 immunohistochemistry is growing in importance.[13][35][39]
Gynecologic cancers
p16 is a widely used immunohistochemical marker in gynecologic pathology. Strong and diffuse cytoplasmic and nuclear expression of p16 in squamous cell carcinomas (SCC) of the female genital tract is strongly associated with high-risk human papilloma virus (HPV) infection and neoplasms of cervical origin. The majority of SCCs of uterine cervix express p16. However, p16 can be expressed in other neoplasms and in several normal human tissues.[40]
Urinary bladder SCCs
More than a third of urinary bladder SCCs express p16. SCCs of urinary bladder express p16 independent of gender. p16 immunohistochemical expression alone cannot be used to discriminate between SCCs arising from uterine cervix versus urinary bladder.[40]
Role in cellular senescence
Concentrations of p16INK4a increase dramatically as tissue ages. p16INK4a, along with senescence-associated beta-galactosidase, is regarded to be a biomarker of cellular senescence.[41] Therefore, p16INK4a could potentially be used as a blood test that measures how fast the body's tissues are aging at a molecular level.[42] Notably, a recent survey of cellular senescence induced by multiple treatments to several cell lines does not identify p16 as belonging to a "core signature" of senescence markers.[43]
It has been used as a target to delay some aging changes in mice.[44]
Role in neurogenesis
Increasing p16INK4a expression during aging is associated with reduced progenitor functions from the subventricular zone, which generates throughout life new neurons migrating to the olfactory bulb, thereby reducing olfactory neurogenesis.[45] Deletion of p16INK4a does not affect neurogenesis in the other adult neurogenic niche, the dentate gyrus of the hippocampus.[45] However, recently, it has been demonstrated that p16INK4a protects from depletion after a powerful proneurogenic stimulus - i.e., running - also stem and progenitor cells of the aged dentate gyrus.[46] In fact, after deletion of p16INK4a, stem cells of the dentate gyrus are greatly activated by running, while, in wild-type p16INK4a dentate gyrus stem cells are not affected by running.[46] Therefore, p16Ink4a plays a role in the maintenance of dentate gyrus stem cells after stimulus, by keeping a reserve of their self-renewal capacity during aging. Since the dentate gyrus plays a key role in spatial and contextual memory formation, p16INK4a is implicated in the maintenance of cognitive functions during aging.
Discovery
Researchers Manuel Serrano, Gregory J. Hannon and David Beach discovered p16 in 1993 and correctly characterized the protein as a cyclin-dependent kinase inhibitor.
Role in carcinogenesis
Since its discovery, p16 has become significant in the field of cancer research. The protein was suspected to be involved in carcinogenesis due to the observation that mutation or deletion in the gene was implicated in human cancer cell lines. The detection of p16 inactivation in familial melanoma supplied further evidence. p16 deletion, mutation, hypermethylation, or overexpression is now associated with various cancers. Whether mutations in p16 can be considered to be driver mutations requires further investigation.[17]
Interactions
p16 has been shown to interact with:
See also
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000147889 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000044303 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- 1 2 "Entrez Gene: CDKN2A cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4)".
- ↑ Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA (April 1994). "Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers". Nature. 368 (6473): 753–756. Bibcode:1994Natur.368..753N. doi:10.1038/368753a0. PMID 8152487. S2CID 13199401.
- ↑ Stone S, Jiang P, Dayananth P, Tavtigian SV, Katcher H, Parry D, et al. (July 1995). "Complex structure and regulation of the P16 (MTS1) locus". Cancer Research. 55 (14): 2988–2994. PMID 7606716.
- 1 2 3 Serrano M, Hannon GJ, Beach D (December 1993). "A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4". Nature. 366 (6456): 704–707. Bibcode:1993Natur.366..704S. doi:10.1038/366704a0. PMID 8259215. S2CID 4368128.
- ↑ Hamosh, Ada. "Cyclin-dependent kinase inhibitor 2A; CDKN2A". OMIM. Retrieved 10 December 2013.
- ↑ "Molecular biology of cancer", Oxford University Press, 2005, ISBN 978-0-19-926472-8, Section 5.3
- ↑ Roussel MF (September 1999). "The INK4 family of cell cycle inhibitors in cancer". Oncogene. 18 (38): 5311–5317. doi:10.1038/sj.onc.1202998. PMID 10498883.
- ↑ Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE (September 2006). "p16INK4a induces an age-dependent decline in islet regenerative potential". Nature. 443 (7110): 453–457. Bibcode:2006Natur.443..453K. doi:10.1038/nature05092. PMID 16957737. S2CID 4402013.
- 1 2 3 4 Rayess H, Wang MB, Srivatsan ES (April 2012). "Cellular senescence and tumor suppressor gene p16". International Journal of Cancer. 130 (8): 1715–1725. doi:10.1002/ijc.27316. PMC 3288293. PMID 22025288.
- ↑ Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G (March 1996). "Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence". Molecular and Cellular Biology. 16 (3): 859–867. doi:10.1128/mcb.16.3.859. PMC 231066. PMID 8622687.
- ↑ Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. (November 2002). "Role of histone H3 lysine 27 methylation in Polycomb-group silencing". Science. 298 (5595): 1039–1043. Bibcode:2002Sci...298.1039C. doi:10.1126/science.1076997. PMID 12351676. S2CID 6265267.
- ↑ Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. (March 2007). "The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells". Genes & Development. 21 (5): 525–530. doi:10.1101/gad.415507. PMC 1820894. PMID 17344414.
- 1 2 Liggett WH, Sidransky D (March 1998). "Role of the p16 tumor suppressor gene in cancer". Journal of Clinical Oncology. 16 (3): 1197–1206. doi:10.1200/JCO.1998.16.3.1197. PMID 9508208.
- ↑ Rocco JW, Sidransky D (March 2001). "p16(MTS-1/CDKN2/INK4a) in cancer progression". Experimental Cell Research. 264 (1): 42–55. doi:10.1006/excr.2000.5149. PMID 11237522.
- ↑ Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, et al. (September 1994). "Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma". Nature Genetics. 8 (1): 27–32. doi:10.1038/ng0994-27. PMID 7726912. S2CID 23195660.
- ↑ Bartsch D, Shevlin DW, Tung WS, Kisker O, Wells SA, Goodfellow PJ (November 1995). "Frequent mutations of CDKN2 in primary pancreatic adenocarcinomas". Genes, Chromosomes & Cancer. 14 (3): 189–195. doi:10.1002/gcc.2870140306. PMID 8589035. S2CID 22823227.
- ↑ Liu L, Lassam NJ, Slingerland JM, Bailey D, Cole D, Jenkins R, Hogg D (July 1995). "Germline p16INK4A mutation and protein dysfunction in a family with inherited melanoma". Oncogene. 11 (2): 405–412. PMID 7624155.
- ↑ Helgadottir H, Höiom V, Jönsson G, Tuominen R, Ingvar C, Borg A, et al. (August 2014). "High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families". Journal of Medical Genetics. 51 (8): 545–552. doi:10.1136/jmedgenet-2014-102320. PMC 4112445. PMID 24935963.
- ↑ Igaki H, Sasaki H, Kishi T, Sakamoto H, Tachimori Y, Kato H, et al. (September 1994). "Highly frequent homozygous deletion of the p16 gene in esophageal cancer cell lines". Biochemical and Biophysical Research Communications. 203 (2): 1090–1095. doi:10.1006/bbrc.1994.2294. PMID 8093026.
- ↑ Puig-Butille JA, Escámez MJ, Garcia-Garcia F, Tell-Marti G, Fabra À, Martínez-Santamaría L, et al. (March 2014). "Capturing the biological impact of CDKN2A and MC1R genes as an early predisposing event in melanoma and non melanoma skin cancer". Oncotarget. 5 (6): 1439–1451. doi:10.18632/oncotarget.1444. PMC 4039222. PMID 24742402.
- ↑ Khor GH, Froemming GR, Zain RB, Abraham MT, Omar E, Tan SK, et al. (2013). "DNA methylation profiling revealed promoter hypermethylation-induced silencing of p16, DDAH2 and DUSP1 in primary oral squamous cell carcinoma". International Journal of Medical Sciences. 10 (12): 1727–1739. doi:10.7150/ijms.6884. PMC 3805925. PMID 24155659.
- ↑ Demokan S, Chuang A, Suoğlu Y, Ulusan M, Yalnız Z, Califano JA, Dalay N (October 2012). "Promoter methylation and loss of p16(INK4a) gene expression in head and neck cancer". Head & Neck. 34 (10): 1470–1475. doi:10.1002/hed.21949. PMID 22106032. S2CID 11512476.
- ↑ Shaw RJ, Liloglou T, Rogers SN, Brown JS, Vaughan ED, Lowe D, et al. (February 2006). "Promoter methylation of P16, RARbeta, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing". British Journal of Cancer. 94 (4): 561–568. doi:10.1038/sj.bjc.6602972. PMC 2361183. PMID 16449996.
- ↑ Sharma G, Mirza S, Prasad CP, Srivastava A, Gupta SD, Ralhan R (April 2007). "Promoter hypermethylation of p16INK4A, p14ARF, CyclinD2 and Slit2 in serum and tumor DNA from breast cancer patients". Life Sciences. 80 (20): 1873–1881. doi:10.1016/j.lfs.2007.02.026. PMID 17383681.
- ↑ Jabłonowski Z, Reszka E, Gromadzińska J, Wąsowicz W, Sosnowski M (June 2011). "Hypermethylation of p16 and DAPK promoter gene regions in patients with non-invasive urinary bladder cancer". Archives of Medical Science. 7 (3): 512–516. doi:10.5114/aoms.2011.23421. PMC 3258754. PMID 22295037.
- ↑ Xu R, Wang F, Wu L, Wang J, Lu C (January 2013). "A systematic review of hypermethylation of p16 gene in esophageal cancer". Cancer Biomarkers. 13 (4): 215–226. doi:10.3233/CBM-130355. PMID 24240582.
- ↑ Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tân PF, et al. (July 2010). "Human papillomavirus and survival of patients with oropharyngeal cancer". The New England Journal of Medicine. 363 (1): 24–35. doi:10.1056/NEJMoa0912217. PMC 2943767. PMID 20530316.
- ↑ Lydiatt WM, Patel SG, O'Sullivan B, Brandwein MS, Ridge JA, Migliacci JC, et al. (March 2017). "Head and Neck cancers-major changes in the American Joint Committee on cancer eighth edition cancer staging manual". CA. 67 (2): 122–137. doi:10.3322/caac.21389. PMID 28128848.
- ↑ Mehanna H, Taberna M, von Buchwald C, Tous S, Brooks J, Mena M, et al. (March 2023). "Prognostic implications of p16 and HPV discordance in oropharyngeal cancer (HNCIG-EPIC-OPC): a multicentre, multinational, individual patient data analysis". The Lancet. Oncology. 24 (3): 239–251. doi:10.1016/s1470-2045(23)00013-x. hdl:2445/198366. PMID 36796393. S2CID 256860640.
- ↑ "Testing for both HPV and p16 can give more precise prognoses in oropharyngeal (throat) cancer". NIHR Evidence. 2023. doi:10.3310/nihrevidence_59749. S2CID 261791439.
- 1 2 Oguejiofor KK, Hall JS, Mani N, Douglas C, Slevin NJ, Homer J, et al. (November 2013). "The prognostic significance of the biomarker p16 in oropharyngeal squamous cell carcinoma". Clinical Oncology. 25 (11): 630–638. doi:10.1016/j.clon.2013.07.003. PMID 23916365.
- ↑ Balgkouranidou I, Liloglou T, Lianidou ES (February 2013). "Lung cancer epigenetics: emerging biomarkers". Biomarkers in Medicine. 7 (1): 49–58. doi:10.2217/bmm.12.111. PMID 23387484.
- ↑ Sinha P, Thorstad WT, Nussenbaum B, Haughey BH, Adkins DR, Kallogjeri D, Lewis JS (January 2014). "Distant metastasis in p16-positive oropharyngeal squamous cell carcinoma: a critical analysis of patterns and outcomes". Oral Oncology. 50 (1): 45–51. doi:10.1016/j.oraloncology.2013.10.007. PMC 3942323. PMID 24211084.
- ↑ Hwang H, Tse C, Rodriguez S, Gown A, Churg A (May 2014). "p16 FISH deletion in surface epithelial mesothelial proliferations is predictive of underlying invasive mesothelioma". The American Journal of Surgical Pathology. 38 (5): 681–688. doi:10.1097/PAS.0000000000000176. PMID 24503757. S2CID 28068784.
- ↑ Dreyer JH, Hauck F, Oliveira-Silva M, Barros MH, Niedobitek G (April 2013). "Detection of HPV infection in head and neck squamous cell carcinoma: a practical proposal". Virchows Archiv. 462 (4): 381–389. doi:10.1007/s00428-013-1393-5. PMID 23503925. S2CID 7469046.
- 1 2 Cioffi-Lavina M, Chapman-Fredricks J, Gomez-Fernandez C, Ganjei-Azar P, Manoharan M, Jorda M (July 2010). "P16 expression in squamous cell carcinomas of cervix and bladder". Applied Immunohistochemistry & Molecular Morphology. 18 (4): 344–347. doi:10.1097/PAI.0b013e3181d2bbd7. PMID 20571342. S2CID 5065484.
- ↑ Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. (July 2016). "Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells". Aging. 8 (7): 1294–1315. doi:10.18632/aging.100991. PMC 4993332. PMID 27391570.
- ↑ Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG, et al. (August 2009). "Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging". Aging Cell. 8 (4): 439–448. doi:10.1111/j.1474-9726.2009.00489.x. PMC 2752333. PMID 19485966.
- ↑ Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M (September 2017). "Unmasking Transcriptional Heterogeneity in Senescent Cells". Current Biology. 27 (17): 2652–2660.e4. doi:10.1016/j.cub.2017.07.033. PMC 5788810. PMID 28844647.
- ↑ Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. (November 2011). "Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders". Nature. 479 (7372): 232–236. Bibcode:2011Natur.479..232B. doi:10.1038/nature10600. PMC 3468323. PMID 22048312.
- 1 2 Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, et al. (September 2006). "Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing". Nature. 443 (7110): 448–452. Bibcode:2006Natur.443..448M. doi:10.1038/nature05091. PMC 2586960. PMID 16957738.
- 1 2 Micheli L, D'Andrea G, Ceccarelli M, Ferri A, Scardigli R, Tirone F (2019). "p16Ink4a Prevents the Activation of Aged Quiescent Dentate Gyrus Stem Cells by Physical Exercise". Frontiers in Cellular Neuroscience. 13: 10. doi:10.3389/fncel.2019.00010. PMC 6374340. PMID 30792628.
- ↑ Zhao L, Samuels T, Winckler S, Korgaonkar C, Tompkins V, Horne MC, Quelle DE (January 2003). "Cyclin G1 has growth inhibitory activity linked to the ARF-Mdm2-p53 and pRb tumor suppressor pathways". Molecular Cancer Research. 1 (3): 195–206. PMID 12556559.
- 1 2 Li J, Melvin WS, Tsai MD, Muscarella P (April 2004). "The nuclear protein p34SEI-1 regulates the kinase activity of cyclin-dependent kinase 4 in a concentration-dependent manner". Biochemistry. 43 (14): 4394–4399. CiteSeerX 10.1.1.386.140. doi:10.1021/bi035601s. PMID 15065884.
- 1 2 Sugimoto M, Nakamura T, Ohtani N, Hampson L, Hampson IN, Shimamoto A, et al. (November 1999). "Regulation of CDK4 activity by a novel CDK4-binding protein, p34(SEI-1)". Genes & Development. 13 (22): 3027–3033. doi:10.1101/gad.13.22.3027. PMC 317153. PMID 10580009.
- ↑ Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, et al. (2007). "Large-scale mapping of human protein-protein interactions by mass spectrometry". Molecular Systems Biology. 3: 89. doi:10.1038/msb4100134. PMC 1847948. PMID 17353931.
- 1 2 Fåhraeus R, Paramio JM, Ball KL, Laín S, Lane DP (January 1996). "Inhibition of pRb phosphorylation and cell-cycle progression by a 20-residue peptide derived from p16CDKN2/INK4A". Current Biology. 6 (1): 84–91. doi:10.1016/S0960-9822(02)00425-6. PMID 8805225. S2CID 23024663.
- ↑ Coleman KG, Wautlet BS, Morrissey D, Mulheron J, Sedman SA, Brinkley P, et al. (July 1997). "Identification of CDK4 sequences involved in cyclin D1 and p16 binding". The Journal of Biological Chemistry. 272 (30): 18869–18874. doi:10.1074/jbc.272.30.18869. PMID 9228064.
- ↑ Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP (September 1998). "Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a". Nature. 395 (6699): 237–243. Bibcode:1998Natur.395..237R. doi:10.1038/26155. PMID 9751050. S2CID 204997058.
- ↑ Kaldis P, Ojala PM, Tong L, Mäkelä TP, Solomon MJ (December 2001). "CAK-independent activation of CDK6 by a viral cyclin". Molecular Biology of the Cell. 12 (12): 3987–3999. doi:10.1091/mbc.12.12.3987. PMC 60770. PMID 11739795.
- 1 2 Ivanchuk SM, Mondal S, Rutka JT (June 2008). "p14ARF interacts with DAXX: effects on HDM2 and p53". Cell Cycle. 7 (12): 1836–1850. doi:10.4161/cc.7.12.6025. PMID 18583933.
- 1 2 Rizos H, Diefenbach E, Badhwar P, Woodruff S, Becker TM, Rooney RJ, Kefford RF (February 2003). "Association of p14ARF with the p120E4F transcriptional repressor enhances cell cycle inhibition". The Journal of Biological Chemistry. 278 (7): 4981–4989. doi:10.1074/jbc.M210978200. PMID 12446718.
- 1 2 3 Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y (December 2003). "Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway". Molecular and Cellular Biology. 23 (23): 8902–8912. doi:10.1128/MCB.23.23.8902-8912.2003. PMC 262682. PMID 14612427.
- 1 2 Zhang Y, Xiong Y, Yarbrough WG (March 1998). "ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways". Cell. 92 (6): 725–734. doi:10.1016/S0092-8674(00)81401-4. PMID 9529249. S2CID 334187.
- ↑ Clark PA, Llanos S, Peters G (July 2002). "Multiple interacting domains contribute to p14ARF mediated inhibition of MDM2". Oncogene. 21 (29): 4498–4507. doi:10.1038/sj.onc.1205558. PMID 12085228.
- ↑ Pomerantz J, Schreiber-Agus N, Liégeois NJ, Silverman A, Alland L, Chin L, et al. (March 1998). "The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53". Cell. 92 (6): 713–723. doi:10.1016/S0092-8674(00)81400-2. PMID 9529248. S2CID 17190271.
- ↑ Vivo M, Calogero RA, Sansone F, Calabrò V, Parisi T, Borrelli L, et al. (April 2001). "The human tumor suppressor arf interacts with spinophilin/neurabin II, a type 1 protein-phosphatase-binding protein". The Journal of Biological Chemistry. 276 (17): 14161–14169. doi:10.1074/jbc.M006845200. PMID 11278317.
External links
- Genes,+p16 at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- CDKN2A human gene location in the UCSC Genome Browser.
- CDKN2A human gene details in the UCSC Genome Browser.
PDB gallery | |
|---|---|
|