The integrated stress response is a cellular stress response conserved in eukaryotic cells that downregulates protein synthesis and upregulates specific genes in response to internal or environmental stresses.[1]
Background
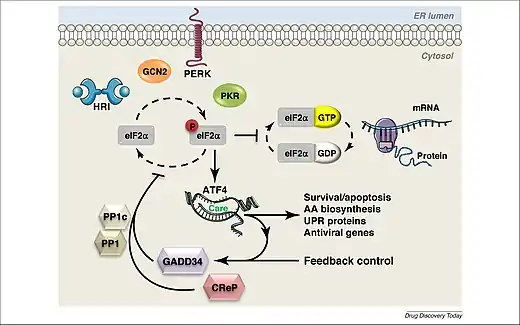
The integrated stress response can be triggered within a cell due to either extrinsic or intrinsic conditions. Extrinsic factors include hypoxia, amino acid deprivation, glucose deprivation, viral infection and presence of oxidants. The main intrinsic factor is endoplasmic reticulum stress due to the accumulation of unfolded proteins. It has also been observed that the integrated stress response may trigger due to oncogene activation. The integrated stress response will either cause the expression of genes that fix the damage in the cell due to the stressful conditions, or it will cause a cascade of events leading to apoptosis, which occurs when the cell cannot be brought back into homeostasis.[1]
eIF2 protein complex
Stress signals can cause protein kinases, known as EIF-2 kinases, to phosphorylate the α subunit of a protein complex called translation initiation factor 2 (eIF2), resulting in the gene ATF4 being turned on, which will further affect gene expression.[1] eIF2 consists of three subunits: eIF2α, eIF2β and eIF2γ. eIF2α contains two binding sites, one for phosphorylation and one for RNA binding.[1] The kinases work to phosphorylate serine 51 on the α subunit, which is a reversible action.[2] In a cell experiencing normal conditions, eIF2 aids in the initiation of mRNA translation and recognizing the AUG start codon.[1] However, once eIF2α is phosphorylated, the complex’s activity reduces, causing reduction in translation initiation and protein synthesis, while promoting expression of the ATF4 gene.[2]
Protein kinases
There are four known mammalian protein kinases that phosphorylate eIF2α, including PKR-like ER kinase (PERK, EIF2AK3), heme-regulated eIF2α kinase (HRI, EIF2AK1), general control non-depressible 2 (GCN2, EIF2AK4) and double stranded RNA dependent protein kinase (PKR, EIF2AK2).[1][3]
PERK

PERK (encoded in humans by the gene EIF2AK3) responds mainly to endoplasmic reticulum stress and has two modes of activation.[1][2] This kinase has a unique luminal domain that plays a role in activation. The classical model of activation states that the luminal domain is normally bound to 78-kDa glucose-regulated protein (GRP78). Once there is a buildup of unfolded proteins, GRP78 dissociates from the luminal domain. This causes PERK to dimerize, leading to autophosphorylation and activation. The activated PERK kinase will then phosphorylate eIF2α, causing a cascade of events. Thus, the activation of this kinase is dependent on the aggregation of unfolded proteins in the endoplasmic reticulum. PERK has also been observed to activate in response to activity of the proto-oncogene MYC. This activation causes ATF4 expression, resulting in tumorigenesis and cellular transformation.[1]

HRI
HRI (encoded in humans by the gene EIF2AK1) also dimerizes in order to autophosphorylate and activate. This activation is dependent on the presence of heme. HRI has two domains that heme may bind to, including one on the N-terminus and one on the kinase insertion domain. The presence of heme causes a disulfide bond to form between the monomers of HRI, resulting in the structure of an inactive dimer. However, when heme is absent, HRI monomers form an active dimer through non-covalent interactions. Therefore, the activation of this kinase is dependent on heme deficiency. HRI activation can also occur due to other stressors such as heat shock, osmotic stress and proteasome inhibition. Activation of HRI in response to these stressors does not depend on heme, but rather relies on the help of two heat shock proteins (HSP90 and HSP70). HRI is mainly found in the precursors of red blood cells, and has been observed to increase during erythropoiesis.[1]

GCN2
GCN2 (encoded in humans by the gene EIF2AK4) is activated as a result of amino acid deprivation. The mechanisms regarding this activation are still being researched; however, one mechanism has been studied in yeast.[1] It was observed that GCN2 binds to uncharged/deacylated tRNA which causes a conformational change, resulting in dimerization.[2] Dimerization then causes autophosphorylation and activation.[2] Other stressors have also been reported to activate GCN2. GCN2 activation was observed in glucose deprived tumor cells, although it was suggested that it was an indirect effect due to cells using amino acids as an alternate energy source.[1] In mouse embryonic fibroblast cells and human keratinocytes, GCN2 was activated due to UV light exposure.[4][5] The pathways for this activation require further research, although multiple models have been proposed, including crosslinking between GCN2 and tRNA.[1]
PKR
PKR (encoded in humans by the gene EIF2AK2) activation is mainly dependent on the presence of double-stranded RNA during a viral infection. dsRNA causes PKR to form dimers, resulting in autophosphorylation and activation.[1] Once activated, PKR will phosphorylate eIF2α which causes a cascade of events that result in viral and host protein synthesis being inhibited. Other stressors that cause the activation of PKR include oxidative stress, endoplasmic reticulum stress, growth factor deprivation and bacterial infection. Caspase activity early on in apoptosis has also been observed to trigger activation of PKR. However, these stressors differ in that they activate PKR without using dsRNA.[1]
ATF4
When a cell is subjected to stressful conditions, the ATF4 gene is expressed.[1] The ATF4 transcription factor has the ability to form dimers with many different proteins that influence gene expression and cell fate. ATF4 binds to C/EBP‐ATF response element (CARE) sequences which work together to increase the transcription of stress-responsive genes. However, when undergoing amino acid starvation, the sequences will act as amino acid response elements instead.[1]
ATF4 will work together with other transcription factors, such as CHOP and ATF3, by forming homodimers or heterodimers, resulting in numerous observed effects.[3] The proteins that ATF4 interacts with determines the outcome of the cell during the integrated stress response.[1] For example, ATF4 and ATF3 work to establish homeostasis inside of the cell following stressful conditions.[3] On the other hand, ATF4 and CHOP work together to induce cell death, as well as regulating amino acid biosynthesis, transport and metabolic processes. The presence of a leucine zipper domain (bZIP) allows ATF4 to work together with many other proteins, thus creating specific responses to different types of stressors. When a cell is undergoing the stress of hypoxia, ATF4 will interact with PHD1 and PHD3 to decrease its transcriptional activity. In addition, when a cell is undergoing amino acid starvation or endoplasmic reticulum stress, TRIP3 also interacts with ATF4 to decrease activity.[1]
One result of ATF4 and stress-response proteins expression is the induction of autophagy.[6] During this process, the cell forms autophagosomes, or double membraned vesicles, that allow for transportation of material throughout the cell.[6] These autophagosomes can carry unneeded organelles and proteins, as well as damaged or harmful components in an attempt by the cell to maintain homeostasis.[6]
Termination of integrated stress response
In order to terminate the integrated stress response, dephosphorylation of eIF2α is required. The protein phosphatase 1 complex (PP1) aids in the dephosphorylation of eIF2α. This complex contains a PP1 catalytic subunit as well as two regulatory subunits. This complex is negatively regulated by two proteins: growth arrest and DNA damage‐inducible protein (GADD34), also known as PPP1R15A, or constitutive repressor of eIF2α phosphorylation (CReP), also known as PPP1R15B. CReP acts to keep levels of eIF2α phosphorylation low in cells under normal conditions. GADD34 is produced in response to ATF4 and works to increase dephosphorylation of eIF2α. The dephosphorylation of eIF2α results in the return of normal protein synthesis and cellular function. However, dephosphorylation of eIF2α can also facilitate the production of death-inducing proteins in cases where the cell is so severely damaged that normal functioning cannot be restored.[1]
Mutations affecting integrated stress response
Mutations that affect the functioning of the integrated stress response may have debilitating effects on cells. For example, cells lacking the ATF4 gene are unable to elicit proper gene expression in response to stressors. This results in cells exhibiting issues with amino acid transport, glutathione biosynthesis and oxidative stress resistance. When a mutation inhibits the functioning of PERK, endogenous peroxides accumulate when the cell experiences endoplasmic reticulum stress.[1] In mice and humans lacking PERK, there have been observed destruction of secretory cells undergoing high endoplasmic reticulum stress.[2]
See also
- ISRIB, integrated stress response inhibitor
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 Pakos‐Zebrucka, Karolina; Koryga, Izabela; Mnich, Katarzyna; Ljujic, Mila; Samali, Afshin; Gorman, Adrienne M (October 2016). "The integrated stress response". EMBO Reports. 17 (10): 1374–1395. doi:10.15252/embr.201642195. PMC 5048378. PMID 27629041.
- 1 2 3 4 5 6 Harding, Heather P.; Zhang, Yuhong; Zeng, Huiquing; Novoa, Isabel; Lu, Phoebe D.; Calfon, Marcella; Sadri, Navid; Yun, Chi; Popko, Brian; Paules, Richard; Stojdl, David F.; Bell, John C.; Hettmann, Thore; Leiden, Jeffrey M.; Ron, David (March 2003). "An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress". Molecular Cell. 11 (3): 619–33. doi:10.1016/S1097-2765(03)00105-9. PMID 12667446.
- 1 2 3 Wang, Cheng; Tan, Zhijia; Niu, Ben; Tsang, Kwok Yeung; Tai, Andrew; Chan, Wilson C W; Lo, Rebecca L K; Leung, Keith K H; Dung, Nelson W F; Itoh, Nobuyuki; Zhang, Michael Q; Chan, Danny; Cheah, Kathryn Song Eng (19 July 2018). "Inhibiting the integrated stress response pathway prevents aberrant chondrocyte differentiation thereby alleviating chondrodysplasia". eLife. 7. doi:10.7554/eLife.37673. PMC 6053305. PMID 30024379.
- ↑ Ovchinnikova, GA; Pigina, TV (February 1975). "[The use of sigetin in the therapy of fetal growth retardation in the rabbit caused by uterine ischemia]". Akusherstvo I Ginekologiia (2): 58–60. PMID 1217635.
- ↑ Lu, Wei; László, Csaba F.; Miao, Zhixin; Chen, Hao; Wu, Shiyong (4 September 2009). "The Role of Nitric-oxide Synthase in the Regulation of UVB Light-induced Phosphorylation of the α Subunit of Eukaryotic Initiation Factor 2". Journal of Biological Chemistry. 284 (36): 24281–24288. doi:10.1074/jbc.M109.008821. PMC 2782021. PMID 19586904.
- 1 2 3 Kroemer, Guido; Mariño, Guillermo; Levine, Beth (October 2010). "Autophagy and the Integrated Stress Response". Molecular Cell. 40 (2): 280–293. doi:10.1016/j.molcel.2010.09.023. PMC 3127250. PMID 20965422.