| Hanhart syndrome | |
|---|---|
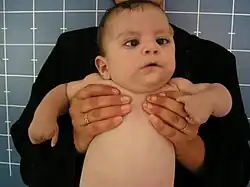 | |
| An infant with Möbius syndrome and Oromandibular-limb hypogenesis syndrome, exhibiting similar symptoms to Hanhart syndrome | |
| Symptoms | Underdevelopment of the mouth, jaw, tongue, and extremities |
| Usual onset | Neonatal, antenatal |
| Duration | Lifelong |
| Causes | Vascular disruption during fetal development |
| Differential diagnosis | Nager acrofacial dysostosis
Johnson Hall Krous syndrome Goldenhar syndrome |
| Frequency | <1/1,000,000, 30 known cases |
Hanhart syndrome[lower-alpha 1] is a broadly classified medical condition consisting of congenital disorders that cause an undeveloped tongue and malformed extremities and fingers. There exist five types of Hanhart syndrome, with the severity and nature of the condition ranging widely on a case-by-case basis. Hanhart syndrome is classified as a rare disease, with approximately 30 known cases having been reported between 1932 and 1991. Early hypotheses believed that the disorder was caused by genetic conditions, with a more recent hypothesis demonstrating that the disorder may be caused by hemorrhagic lesions during prenatal development. The causal mechanism behind this vascular disruption is still unknown.
Discovery and etymology
Hanhart syndrome was first described in 1932 by Ernst Hanhart. The name "Hanhart syndrome" was not used until 1950 when Hanhart described three patients who were born with limb defects and missing tongues.[1] In 1971, the syndrome was more broadly classified as syndromes of oromandibular and limb hypogenesis, and comprised a range of disorders that all share hypoglossia. The name ''hypoglossia-hypodactylia syndrome' was proposed in 1971 as a more accurate name for the disorder, which is used synonymously.[2][3] Due to the wide range of symptoms presented in Hanhart syndrome, the disorder has received many different names throughout its diagnostic history.[4]
Pathophysiology
Hanhart syndrome is part of the larger oromandibular-limb hypogenesis syndrome (OLHS) family of conditions, which are collectively characterized by the underdevelopment of the mouth, jaw, tongue, and extremities.[2][1] Hanhart syndrome is characterized by an underdeveloped tongue (hypoglossia), small mouth (microstomia), smaller than average jaw size (micrognathia), clefting or abnormal attachment of the tongue, missing teeth (mandibular hypodontia), cleft palate, cranial nerve palsies including Möbius syndrome, broad nose, increased distance between the eyes (telecanthus), defects in the lower eyelids, and facial asymmetry.[2][1][5]

Limbs generally present as underdeveloped (hypoplasia), which can vary from missing phalanx bones (hypodactylia) to incomplete adactyly to partial amputation or malformation of the limbs (peromelia). In cases where limb defects are involved, they generally involve all four extremities.[5] Complete loss of the tongue (aglossia) and fingers (adactylia) have not been reported.[3] Less common symptoms include externally visible intestines (gastroschisis) and abnormal fusion of the spleen and gonads (splenogonadal fusion).[5] Supernumerary nipples, microcephaly, and micropenis have also been identified as possible symptoms.[6] Ultrasound examination of the cranial and abdominal regions in patients has found that internal structures are unaffected by Hanhart syndrome.[7] Mental disabilities are uncommon, but feeding and speech deficits have been reported in some cases.[5] The severity of these disabilities can vary greatly from person to person, and patients with the disorder often present some, but not all, of the symptoms.[2][1]
Hanhart syndrome is classified by five types, denoting the symptoms and severity of the condition. Type I involves hypoglossia and partial aglossia without any other symptoms. Type II involves hypoglossia with hypomelia or hypodactylia. Type III includes hypoglossia with glossopalatine ankylosis and hypomelia or hypodactyly. Type IV involves fused inter-oral bands with hypoglossia or hypomelia/hypodactyly. Type V involves several co-morbid syndromes alongside Hanhart syndrome, including: Pierre Robin syndrome, Mobius syndrome, and amniotic band syndrome.[3][8]
Causes and prevalence
The causes of Hanhart syndrome and other OLHS conditions is unknown, but both genetic and environment factors have been proposed.[9] Hanhart syndrome follows an autosomal dominant inheritance pattern, has a population prevalence of <1/1,000,000, and due to its rarity has been classified as a rare disease.[5][10] Between 1932 and 1991, about 30 case of Hanhart syndrome were reported in the literature.[5]
The prevalence of Hanhart syndrome within consanguineous patients lead to an early hypothesis that a mutation in an autosomal recessive gene led to the condition.[5] To date, no specific genes have been identified.[7] In 1973, a hypothesis that hemorrhagic lesions or blood clots during prenatal development were the cause of Hanhart syndrome was proposed. This hypothesis asserts that vascular lesions result in decreased blood flow to the limbs, tongue, and rarely parts of the brain, causing developmental malformation in the affected parts of the body.[1][5] Animal studies have shown that vascular disruptions in the fourth embryonic week can result in Hanhart syndrome, but the mechanism of this disruption is still unknown.[11] Interactions between the ectodermal and mesodermal germ layers,[4] and the use of meclizine hydrochloride during pregnancy have been proposed as a potential causes.[12] It has also been proposed that Poland syndrome and Hanhart syndrome may have similar causes and represent the spectrum of a larger disorder.[4]
The use of chorionic villous sampling (CVS) has also been identified as a contributing factor to pathogensis. When CVS is used during the early stages of pregnancy, specifically during the first 10 weeks of amenorrhoea, there is an increased correlation with risk of the fetus developing Hanhart syndrome. Furthermore, the use of CVS is known to introduce vascular damages to the fetus, causing oromandibular limb dysgenesis and other prenatal disorders, giving credence to the vascular disruption hypothesis.[13][5][1]
Diagnosis and treatments
Diagnosis of Hanhart syndrome relies on clinical presentation.[11] Differential diagnosis of Hanhart syndrome must rule out Nager syndrome and acro-facial dysostosis, Johnson Hall Krous syndrome, and Goldenhar syndrome.[11][5] These syndromes are differentiated by the type and severity of facial and limb deformities.[5]
Treatments for Hanhart syndrome vary depending on the specific symptoms per patient.[2] Treatments may include orthopedic or plastic surgery to treat for limb deformities.[5] These can include surgical correction of the tongue, mouth, and jaw regions, as well as the use of prostheses and physical therapy. Speech therapy is another common treatment for facial deformities.[1] Special consideration of the airway is often taken during operations under anesthesia due to craniofacial deformities.[11]
Notable cases
References
- 1 2 3 4 5 6 7 "Hanhart Syndrome - National Organization for Rare Disorders". rarediseases.org. Archived from the original on 2021-09-30. Retrieved 2023-02-25.
- 1 2 3 4 5 "Hanhart syndrome - National Organization for Rare Disorders". rarediseases.org. 2022-06-16. Archived from the original on 2023-02-25. Retrieved 2023-02-25.
- 1 2 3 McKusick VA, Kniffin CL (August 8, 2016). "Hypoglossia-Hypodactylia". omim.org. Archived from the original on 2022-06-17. Retrieved 2023-02-25.
- 1 2 3 Herrmann J, Pallister PD, Gilbert EF, Vieseskul C, Bersu E, Pettersen JC, Opitz JM (April 1976). "Studies of malformation syndromes of man XXXXI B: nosologic studies in the Hanhart and the Möbius syndrome". European Journal of Pediatrics. 122 (1): 19–55. doi:10.1007/BF00445030. PMID 1261566. S2CID 23498856.
- 1 2 3 4 5 6 7 8 9 10 11 12 "Orphanet: Hypoglossia hypodactyly syndrome". www.orpha.net. Archived from the original on 2023-02-25. Retrieved 2023-02-25.
- ↑ Gathwala G, Singh J, Dalal P, Garg A (March 2011). "Hypoglossia-hypodactyly syndrome in a newborn". Journal of Cranio-Maxillo-Facial Surgery. 39 (2): 99–101. doi:10.1016/j.jcms.2010.06.007. PMID 20673638.
- 1 2 Varal IG, Dogan P (2019-04-29). "Hanhart syndrome: hypoglossia-hypodactylia syndrome". Pan African Medical Journal. 32 (1): 213. ISSN 1937-8688. PMC 6620079. PMID 31312325. Archived from the original on 2023-02-26. Retrieved 2023-03-10.
- ↑ Goyal M, Singh A, Singh P, Kapoor S (April 2014). "Hypoglossia-hypodactyly syndrome with short stature - a case report". Journal of Clinical and Diagnostic Research. 8 (4): SD01–SD02. doi:10.7860/JCDR/2014/7809.4281 (inactive 1 August 2023). PMC 4064859. PMID 24959494.
{{cite journal}}: CS1 maint: DOI inactive as of August 2023 (link) - ↑ Bersu ET, Pettersen JC, Charboneau WJ, Opitz JM (April 1976). "Studies of malformation syndromes of man XXXXIA: anatomical studies in the Hanhart syndrome--a pathogenetic hypothesis". European Journal of Pediatrics. 122 (1): 1–17. doi:10.1007/BF00445029. PMID 1261565. S2CID 10440385.
- ↑ "Hanhart syndrome - About the Disease - Genetic and Rare Diseases Information Center". rarediseases.info.nih.gov. Archived from the original on 2023-01-05. Retrieved 2023-02-25.
- 1 2 3 4 Bissonnette, Bruno; Luginbuehl, Igor; Engelhardt, Thomas (2019), "Hypoglossia-Hypodactylia Syndrome", Syndromes: Rapid Recognition and Perioperative Implications (2 ed.), New York, NY: McGraw-Hill Education, archived from the original on 2023-03-10, retrieved 2023-02-27
- ↑ Bökesoy I, Aksüyek C, Deniz E (July 1983). "Oromandibular limb hypogenesis/Hanhart's syndrome: possible drug influence on the malformation". Clinical Genetics. 24 (1): 47–49. doi:10.1111/j.1399-0004.1983.tb00068.x. PMID 6616945. S2CID 43535751.
- ↑ Chen, C. P.; Liu, F. F.; Jan, S. W.; Lin, S. P.; Lan, C. C. (1996-06-14). "CVS-exposed limb deficiency defects with or without other birth defects: presentation of six cases born during a period of nine years". American Journal of Medical Genetics. 63 (3): 447–453. doi:10.1002/(SICI)1096-8628(19960614)63:3<447::AID-AJMG6>3.0.CO;2-O. ISSN 0148-7299. PMID 8737650.
- ↑ Petter O (20 July 2017). "Bodybuilder with one arm and no legs becomes internet sensation". The Independent. Archived from the original on 19 April 2018. Retrieved 19 April 2018.
- ↑ Stump S (October 2016). "'Dead' man living: Disabled teen-turned-Internet star 'lives by inspiring others'". Today. Archived from the original on 2018-04-19. Retrieved 2018-04-19.
Notes
- ↑ Also known as Aglossia adactylia; Hypoglossia-hypodactylia syndrome; Peromelia with micrognathia; and Jussieu syndrome