
Enzyme kinetics is the study of the rates of enzyme-catalysed chemical reactions. In enzyme kinetics, the reaction rate is measured and the effects of varying the conditions of the reaction are investigated. Studying an enzyme's kinetics in this way can reveal the catalytic mechanism of this enzyme, its role in metabolism, how its activity is controlled, and how a drug or a modifier (inhibitor or activator) might affect the rate.
An enzyme (E) is typically a protein molecule that promotes a reaction of another molecule, its substrate (S). This binds to the active site of the enzyme to produce an enzyme-substrate complex ES, and is transformed into an enzyme-product complex EP and from there to product P, via a transition state ES*. The series of steps is known as the mechanism:
- E + S ⇄ ES ⇄ ES* ⇄ EP ⇄ E + P
This example assumes the simplest case of a reaction with one substrate and one product. Such cases exist: for example, a mutase such as phosphoglucomutase catalyses the transfer of a phospho group from one position to another, and isomerase is a more general term for an enzyme that catalyses any one-substrate one-product reaction, such as triosephosphate isomerase. However, such enzymes are not very common, and are heavily outnumbered by enzymes that catalyse two-substrate two-product reactions: these include, for example, the NAD-dependent dehydrogenases such as alcohol dehydrogenase, which catalyses the oxidation of ethanol by NAD+. Reactions with three or four substrates or products are less common, but they exist. There is no necessity for the number of products to be equal to the number of substrates; for example, glyceraldehyde 3-phosphate dehydrogenase has three substrates and two products.
When enzymes bind multiple substrates, such as dihydrofolate reductase (shown right), enzyme kinetics can also show the sequence in which these substrates bind and the sequence in which products are released. An example of enzymes that bind a single substrate and release multiple products are proteases, which cleave one protein substrate into two polypeptide products. Others join two substrates together, such as DNA polymerase linking a nucleotide to DNA. Although these mechanisms are often a complex series of steps, there is typically one rate-determining step that determines the overall kinetics. This rate-determining step may be a chemical reaction or a conformational change of the enzyme or substrates, such as those involved in the release of product(s) from the enzyme.
Knowledge of the enzyme's structure is helpful in interpreting kinetic data. For example, the structure can suggest how substrates and products bind during catalysis; what changes occur during the reaction; and even the role of particular amino acid residues in the mechanism. Some enzymes change shape significantly during the mechanism; in such cases, it is helpful to determine the enzyme structure with and without bound substrate analogues that do not undergo the enzymatic reaction.
Not all biological catalysts are protein enzymes: RNA-based catalysts such as ribozymes and ribosomes are essential to many cellular functions, such as RNA splicing and translation. The main difference between ribozymes and enzymes is that RNA catalysts are composed of nucleotides, whereas enzymes are composed of amino acids. Ribozymes also perform a more limited set of reactions, although their reaction mechanisms and kinetics can be analysed and classified by the same methods.
General principles
.svg.png.webp)

The reaction catalysed by an enzyme uses exactly the same reactants and produces exactly the same products as the uncatalysed reaction. Like other catalysts, enzymes do not alter the position of equilibrium between substrates and products.[1] However, unlike uncatalysed chemical reactions, enzyme-catalysed reactions display saturation kinetics. For a given enzyme concentration and for relatively low substrate concentrations, the reaction rate increases linearly with substrate concentration; the enzyme molecules are largely free to catalyse the reaction, and increasing substrate concentration means an increasing rate at which the enzyme and substrate molecules encounter one another. However, at relatively high substrate concentrations, the reaction rate asymptotically approaches the theoretical maximum; the enzyme active sites are almost all occupied by substrates resulting in saturation, and the reaction rate is determined by the intrinsic turnover rate of the enzyme.[2] The substrate concentration midway between these two limiting cases is denoted by KM. Thus, KM is the substrate concentration at which the reaction velocity is half of the maximum velocity.[2]
The two important properties of enzyme kinetics are how easily the enzyme can be saturated with a substrate, and the maximum rate it can achieve. Knowing these properties suggests what an enzyme might do in the cell and can show how the enzyme will respond to changes in these conditions.
Enzyme assays

Enzyme assays are laboratory procedures that measure the rate of enzyme reactions. Since enzymes are not consumed by the reactions they catalyse, enzyme assays usually follow changes in the concentration of either substrates or products to measure the rate of reaction. There are many methods of measurement. Spectrophotometric assays observe the change in the absorbance of light between products and reactants; radiometric assays involve the incorporation or release of radioactivity to measure the amount of product made over time. Spectrophotometric assays are most convenient since they allow the rate of the reaction to be measured continuously. Although radiometric assays require the removal and counting of samples (i.e., they are discontinuous assays) they are usually extremely sensitive and can measure very low levels of enzyme activity.[3] An analogous approach is to use mass spectrometry to monitor the incorporation or release of stable isotopes as the substrate is converted into product. Occasionally, an assay fails and approaches are essential to resurrect a failed assay.
The most sensitive enzyme assays use lasers focused through a microscope to observe changes in single enzyme molecules as they catalyse their reactions. These measurements either use changes in the fluorescence of cofactors during an enzyme's reaction mechanism, or of fluorescent dyes added onto specific sites of the protein to report movements that occur during catalysis.[4] These studies provide a new view of the kinetics and dynamics of single enzymes, as opposed to traditional enzyme kinetics, which observes the average behaviour of populations of millions of enzyme molecules.[5][6]
An example progress curve for an enzyme assay is shown above. The enzyme produces product at an initial rate that is approximately linear for a short period after the start of the reaction. As the reaction proceeds and substrate is consumed, the rate continuously slows (so long as the substrate is not still at saturating levels). To measure the initial (and maximal) rate, enzyme assays are typically carried out while the reaction has progressed only a few percent towards total completion. The length of the initial rate period depends on the assay conditions and can range from milliseconds to hours. However, equipment for rapidly mixing liquids allows fast kinetic measurements at initial rates of less than one second.[7] These very rapid assays are essential for measuring pre-steady-state kinetics, which are discussed below.
Most enzyme kinetics studies concentrate on this initial, approximately linear part of enzyme reactions. However, it is also possible to measure the complete reaction curve and fit this data to a non-linear rate equation. This way of measuring enzyme reactions is called progress-curve analysis.[8] This approach is useful as an alternative to rapid kinetics when the initial rate is too fast to measure accurately.
The Standards for Reporting Enzymology Data Guidelines provide minimum information required to comprehensively report kinetic and equilibrium data from investigations of enzyme activities including corresponding experimental conditions. The guidelines have been developed to report functional enzyme data with rigor and robustness.
Single-substrate reactions
Enzymes with single-substrate mechanisms include isomerases such as triosephosphateisomerase or bisphosphoglycerate mutase, intramolecular lyases such as adenylate cyclase and the hammerhead ribozyme, an RNA lyase.[9] However, some enzymes that only have a single substrate do not fall into this category of mechanisms. Catalase is an example of this, as the enzyme reacts with a first molecule of hydrogen peroxide substrate, becomes oxidised and is then reduced by a second molecule of substrate. Although a single substrate is involved, the existence of a modified enzyme intermediate means that the mechanism of catalase is actually a ping–pong mechanism, a type of mechanism that is discussed in the Multi-substrate reactions section below.
Michaelis–Menten kinetics


As enzyme-catalysed reactions are saturable, their rate of catalysis does not show a linear response to increasing substrate. If the initial rate of the reaction is measured over a range of substrate concentrations (denoted as [S]), the initial reaction rate () increases as [S] increases, as shown on the right. However, as [S] gets higher, the enzyme becomes saturated with substrate and the initial rate reaches Vmax, the enzyme's maximum rate.

The Michaelis–Menten kinetic model of a single-substrate reaction is shown on the right. There is an initial bimolecular reaction between the enzyme E and substrate S to form the enzyme–substrate complex ES. The rate of enzymatic reaction increases with the increase of the substrate concentration up to a certain level called Vmax; at Vmax, increase in substrate concentration does not cause any increase in reaction rate as there is no more enzyme (E) available for reacting with substrate (S). Here, the rate of reaction becomes dependent on the ES complex and the reaction becomes a unimolecular reaction with an order of zero. Though the enzymatic mechanism for the unimolecular reaction can be quite complex, there is typically one rate-determining enzymatic step that allows this reaction to be modelled as a single catalytic step with an apparent unimolecular rate constant kcat. If the reaction path proceeds over one or several intermediates, kcat will be a function of several elementary rate constants, whereas in the simplest case of a single elementary reaction (e.g. no intermediates) it will be identical to the elementary unimolecular rate constant k2. The apparent unimolecular rate constant kcat is also called turnover number, and denotes the maximum number of enzymatic reactions catalysed per second.
![{\displaystyle {\ce {ES ->[k_{cat}] E + P}}}](../I/299f3433b9ca64a864deef13f572a0127a2d14e0.svg)
The Michaelis–Menten equation[10] describes how the (initial) reaction rate v0 depends on the position of the substrate-binding equilibrium and the rate constant k2.
- (Michaelis–Menten equation)
![{\displaystyle v_{0}={\frac {V_{\max }[{\ce {S}}]}{K_{M}+[{\ce {S}}]}}}](../I/8f2f1d1e9d417b925f380340d6d3581d4006672f.svg)
with the constants
![{\displaystyle {\begin{aligned}K_{M}\ &{\stackrel {\mathrm {def} }{=}}\ {\frac {k_{2}+k_{-1}}{k_{1}}}\approx K_{D}\\V_{\max }\ &{\stackrel {\mathrm {def} }{=}}\ k_{cat}{\ce {[E]}}_{tot}\end{aligned}}}](../I/1714252e04d803899ef6ad5b75c074d0f9ebc50c.svg)
This Michaelis–Menten equation is the basis for most single-substrate enzyme kinetics. Two crucial assumptions underlie this equation (apart from the general assumption about the mechanism only involving no intermediate or product inhibition, and there is no allostericity or cooperativity). The first assumption is the so-called quasi-steady-state assumption (or pseudo-steady-state hypothesis), namely that the concentration of the substrate-bound enzyme (and hence also the unbound enzyme) changes much more slowly than those of the product and substrate and thus the change over time of the complex can be set to zero . The second assumption is that the total enzyme concentration does not change over time, thus . A complete derivation can be found here.
![{\displaystyle d{\ce {[ES]}}/{dt}\;{\overset {!}{=}}\;0}](../I/0eea1ce2a30471bd05b46fe979bb4f12e365b4d5.svg)
![{\displaystyle {\ce {[E]}}_{\text{tot}}={\ce {[E]}}+{\ce {[ES]}}\;{\overset {!}{=}}\;{\text{const}}}](../I/5c082974766078275f236f456e12426c4ea02fc8.svg)
The Michaelis constant KM is experimentally defined as the concentration at which the rate of the enzyme reaction is half Vmax, which can be verified by substituting [S] = KM into the Michaelis–Menten equation and can also be seen graphically. If the rate-determining enzymatic step is slow compared to substrate dissociation (), the Michaelis constant KM is roughly the dissociation constant KD of the ES complex.

If is small compared to then the term and also very little ES complex is formed, thus . Therefore, the rate of product formation is
![{\displaystyle {\ce {[S]}}}](../I/e5909e9989dfe9306325e8dab287928f3c984ee3.svg)

![{\displaystyle [{\ce {S}}]/(K_{M}+[{\ce {S}}])\approx [{\ce {S}}]/K_{M}}](../I/1d1befa5e00217f79ed63dc6ba5c6a15d78d5425.svg)
![{\displaystyle {\ce {[E]_{\rm {tot}}\approx [E]}}}](../I/f6ccda6e543afca4e4287f635c5f1a4931ca93e1.svg)
![{\displaystyle v_{0}\approx {\frac {k_{cat}}{K_{M}}}{\ce {[E][S]}}\qquad \qquad {\text{if }}[{\ce {S}}]\ll K_{M}}](../I/596b2c4659de250ffbd0b65c085402f9fd16735d.svg)
Thus the product formation rate depends on the enzyme concentration as well as on the substrate concentration, the equation resembles a bimolecular reaction with a corresponding pseudo-second order rate constant . This constant is a measure of catalytic efficiency. The most efficient enzymes reach a in the range of 108 – 1010 M−1 s−1. These enzymes are so efficient they effectively catalyse a reaction each time they encounter a substrate molecule and have thus reached an upper theoretical limit for efficiency (diffusion limit); and are sometimes referred to as kinetically perfect enzymes.[11] But most enzymes are far from perfect: the average values of and are about and , respectively.[12]





Direct use of the Michaelis–Menten equation for time course kinetic analysis
The observed velocities predicted by the Michaelis–Menten equation can be used to directly model the time course disappearance of substrate and the production of product through incorporation of the Michaelis–Menten equation into the equation for first order chemical kinetics. This can only be achieved however if one recognises the problem associated with the use of Euler's number in the description of first order chemical kinetics. i.e. e−k is a split constant that introduces a systematic error into calculations and can be rewritten as a single constant which represents the remaining substrate after each time period.[13]
![[S]=[S]_{0}(1-k)^{{t}}\,](../I/93211e467eb88a4ed3ce4b1b8a64f3645c540709.svg)
![[S]=[S]_{0}(1-v/[S]_{0})^{{t}}\,](../I/fc767ed4ec3fb17dbb2b342b438ca22f3a0c5e15.svg)
![[S]=[S]_{0}(1-(V_{{\max }}[S]_{0}/(K_{M}+[S]_{0})/[S]_{0}))^{{t}}\,](../I/550cebc162d8baf678f05a64c8435882eba78bfb.svg)
In 1983 Stuart Beal (and also independently Santiago Schnell and Claudio Mendoza in 1997) derived a closed form solution for the time course kinetics analysis of the Michaelis-Menten mechanism.[14][15] The solution, known as the Schnell-Mendoza equation, has the form:
![{\frac {[S]}{K_{M}}}=W\left[F(t)\right]\,](../I/bbfb88da686a3b0298417f08709f60c89538b35e.svg)
where W[ ] is the Lambert-W function.[16][17] and where F(t) is
![F(t) = \frac{[S]_0}{K_M} \exp\!\left(\frac{[S]_0}{K_M} - \frac{V_\max}{K_M}\,t \right) \,](../I/39737501b38ca63037f8350456c777481706c602.svg)
This equation is encompassed by the equation below, obtained by Berberan-Santos,[18] which is also valid when the initial substrate concentration is close to that of enzyme,
![{\frac {[S]}{K_{M}}}=W\left[F(t)\right]-{\frac {V_{\max }}{k_{{cat}}K_{M}}}\ {\frac {W\left[F(t)\right]}{1+W\left[F(t)\right]}}\,](../I/036e38cefdac7ce988899ea0d4b89f80c0b6e81d.svg)
where W[ ] is again the Lambert-W function.
Linear plots of the Michaelis–Menten equation

The plot of v versus [S] above is not linear; although initially linear at low [S], it bends over to saturate at high [S]. Before the modern era of nonlinear curve-fitting on computers, this nonlinearity could make it difficult to estimate KM and Vmax accurately. Therefore, several researchers developed linearisations of the Michaelis–Menten equation, such as the Lineweaver–Burk plot, the Eadie–Hofstee diagram and the Hanes–Woolf plot. All of these linear representations can be useful for visualising data, but none should be used to determine kinetic parameters, as computer software is readily available that allows for more accurate determination by nonlinear regression methods.[19]
The Lineweaver–Burk plot or double reciprocal plot is a common way of illustrating kinetic data. This is produced by taking the reciprocal of both sides of the Michaelis–Menten equation. As shown on the right, this is a linear form of the Michaelis–Menten equation and produces a straight line with the equation y = mx + c with a y-intercept equivalent to 1/Vmax and an x-intercept of the graph representing −1/KM.
![{\frac {1}{v}}={\frac {K_{{M}}}{V_{{\max }}[{\mbox{S}}]}}+{\frac {1}{V_{\max }}}](../I/f67c173c3e3e8c78da7dc5fa15c3b5ff299e4439.svg)
Naturally, no experimental values can be taken at negative 1/[S]; the lower limiting value 1/[S] = 0 (the y-intercept) corresponds to an infinite substrate concentration, where 1/v=1/Vmax as shown at the right; thus, the x-intercept is an extrapolation of the experimental data taken at positive concentrations. More generally, the Lineweaver–Burk plot skews the importance of measurements taken at low substrate concentrations and, thus, can yield inaccurate estimates of Vmax and KM.[20] A more accurate linear plotting method is the Eadie–Hofstee plot. In this case, v is plotted against v/[S]. In the third common linear representation, the Hanes–Woolf plot, [S]/v is plotted against [S]. In general, data normalisation can help diminish the amount of experimental work and can increase the reliability of the output, and is suitable for both graphical and numerical analysis.[21]
Practical significance of kinetic constants
The study of enzyme kinetics is important for two basic reasons. Firstly, it helps explain how enzymes work, and secondly, it helps predict how enzymes behave in living organisms. The kinetic constants defined above, KM and Vmax, are critical to attempts to understand how enzymes work together to control metabolism.
Making these predictions is not trivial, even for simple systems. For example, oxaloacetate is formed by malate dehydrogenase within the mitochondrion. Oxaloacetate can then be consumed by citrate synthase, phosphoenolpyruvate carboxykinase or aspartate aminotransferase, feeding into the citric acid cycle, gluconeogenesis or aspartic acid biosynthesis, respectively. Being able to predict how much oxaloacetate goes into which pathway requires knowledge of the concentration of oxaloacetate as well as the concentration and kinetics of each of these enzymes. This aim of predicting the behaviour of metabolic pathways reaches its most complex expression in the synthesis of huge amounts of kinetic and gene expression data into mathematical models of entire organisms. Alternatively, one useful simplification of the metabolic modelling problem is to ignore the underlying enzyme kinetics and only rely on information about the reaction network's stoichiometry, a technique called flux balance analysis.[22][23]
Michaelis–Menten kinetics with intermediate
One could also consider the less simple case
![{\displaystyle {\ce {{E}+S<=>[k_{1}][k_{-1}]ES->[k_{2}]EI->[k_{3}]{E}+P}}}](../I/5cd1904bee27689d5df0933e40a4b01631243041.svg)
where a complex with the enzyme and an intermediate exists and the intermediate is converted into product in a second step. In this case we have a very similar equation[24]
![{\displaystyle v_{0}=k_{cat}{\frac {{\ce {[S] [E]_0}}}{K_{M}^{\prime }+{\ce {[S]}}}}}](../I/d90d4c1b92e79705a9ecf0f8615982c0bc91f4a3.svg)
but the constants are different

We see that for the limiting case , thus when the last step from is much faster than the previous step, we get again the original equation. Mathematically we have then and .




Multi-substrate reactions
Multi-substrate reactions follow complex rate equations that describe how the substrates bind and in what sequence. The analysis of these reactions is much simpler if the concentration of substrate A is kept constant and substrate B varied. Under these conditions, the enzyme behaves just like a single-substrate enzyme and a plot of v by [S] gives apparent KM and Vmax constants for substrate B. If a set of these measurements is performed at different fixed concentrations of A, these data can be used to work out what the mechanism of the reaction is. For an enzyme that takes two substrates A and B and turns them into two products P and Q, there are two types of mechanism: ternary complex and ping–pong.
Ternary-complex mechanisms

In these enzymes, both substrates bind to the enzyme at the same time to produce an EAB ternary complex. The order of binding can either be random (in a random mechanism) or substrates have to bind in a particular sequence (in an ordered mechanism). When a set of v by [S] curves (fixed A, varying B) from an enzyme with a ternary-complex mechanism are plotted in a Lineweaver–Burk plot, the set of lines produced will intersect.
Enzymes with ternary-complex mechanisms include glutathione S-transferase,[25] dihydrofolate reductase[26] and DNA polymerase.[27] The following links show short animations of the ternary-complex mechanisms of the enzymes dihydrofolate reductase[β] and DNA polymerase[γ].
Ping–pong mechanisms
![{\displaystyle {\ce {\overset {}{E->[{\ce {A \atop \downarrow }}]EA<=>E^{\ast }P->[{\ce {P \atop \uparrow }}]E^{\ast }->[{\ce {B \atop \downarrow }}]E^{\ast }B<=>EQ->[{\ce {Q \atop \uparrow }}]E}}}}](../I/f9b768dbbd547267c22748f29590cb0a639375de.svg)
As shown on the right, enzymes with a ping-pong mechanism can exist in two states, E and a chemically modified form of the enzyme E*; this modified enzyme is known as an intermediate. In such mechanisms, substrate A binds, changes the enzyme to E* by, for example, transferring a chemical group to the active site, and is then released. Only after the first substrate is released can substrate B bind and react with the modified enzyme, regenerating the unmodified E form. When a set of v by [S] curves (fixed A, varying B) from an enzyme with a ping–pong mechanism are plotted in a Lineweaver–Burk plot, a set of parallel lines will be produced. This is called a secondary plot.
Enzymes with ping–pong mechanisms include some oxidoreductases such as thioredoxin peroxidase,[28] transferases such as acylneuraminate cytidylyltransferase[29] and serine proteases such as trypsin and chymotrypsin.[30] Serine proteases are a very common and diverse family of enzymes, including digestive enzymes (trypsin, chymotrypsin, and elastase), several enzymes of the blood clotting cascade and many others. In these serine proteases, the E* intermediate is an acyl-enzyme species formed by the attack of an active site serine residue on a peptide bond in a protein substrate. A short animation showing the mechanism of chymotrypsin is linked here.[δ]
Reversible catalysis and the Haldane equation
External factors may limit the ability of an enzyme to catalyse a reaction in both directions (whereas the nature of a catalyst in itself means that it cannot catalyse just one direction, according to the principle of microscopic reversibility). We consider the case of an enzyme that catalyses the reaction in both directions:
![{\displaystyle {\ce {{E}+{S}<=>[k_{1}][k_{-1}]ES<=>[k_{2}][k_{-2}]{E}+{P}}}}](../I/fe362b85b97b0614f6139dd0be2e8389a2c3b63e.svg)
The steady-state, initial rate of the reaction is
![{\displaystyle v_{0}={\frac {d\,[{\rm {P}}]}{dt}}={\frac {(k_{1}k_{2}\,[{\rm {S}}]-k_{-1}k_{-2}[{\rm {P}}])[{\rm {E}}]_{0}}{k_{-1}+k_{2}+k_{1}\,[{\rm {S}}]+k_{-2}\,[{\rm {P}}]}}}](../I/01297c24d81598b7438d064174294408dacc5e60.svg)
is positive if the reaction proceed in the forward direction () and negative otherwise.

Equilibrium requires that , which occurs when . This shows that thermodynamics forces a relation between the values of the 4 rate constants.

![{\displaystyle {\frac {[{\rm {P}}]_{\rm {eq}}}{[{\rm {S}}]_{\rm {eq}}}}={\frac {k_{1}k_{2}}{k_{-1}k_{-2}}}=K_{\rm {eq}}}](../I/4f0ca30bfb4f852fcc02a12f729bcbd3adc500d6.svg)
The values of the forward and backward maximal rates, obtained for , , and , , respectively, are and , respectively. Their ratio is not equal to the equilibrium constant, which implies that thermodynamics does not constrain the ratio of the maximal rates. This explains that enzymes can be much "better catalysts" (in terms of maximal rates) in one particular direction of the reaction.[31]
![{\displaystyle [{\rm {S}}]\rightarrow \infty }](../I/c3e69387ec816688807461db5902b440a6a65ed0.svg)
![{\displaystyle [{\rm {P}}]=0}](../I/93c498e9bfae3b2d0f1378c8a1a15031f4002279.svg)
![{\displaystyle [{\rm {S}}]=0}](../I/53ac89660d3f5c0ad06eac244190defc17cd7a61.svg)
![{\displaystyle [{\rm {P}}]\rightarrow \infty }](../I/020c2fe953abd3c508b7bd3a6d97bb29225faeeb.svg)
![{\displaystyle V_{\rm {max}}^{f}=k_{2}{\rm {[E]}}_{tot}}](../I/1834ca7ac4c486ffc3e1c89742f31c266797da98.svg)
![{\displaystyle V_{\rm {max}}^{b}=-k_{-1}{\rm {[E]}}_{tot}}](../I/a09363446309c8c26702430b70bfec463a3b0134.svg)
On can also derive the two Michaelis constants and . The Haldane equation is the relation .


![{\displaystyle K_{\rm {eq}}={\frac {[{\rm {P}}]_{\rm {eq}}}{[{\rm {S}}]_{\rm {eq}}}}={\frac {V_{\rm {max}}^{f}/K_{M}^{S}}{V_{\rm {max}}^{b}/K_{M}^{P}}}}](../I/621e1b38683241916244c9e90f5625f14f1f0088.svg)
Therefore, thermodynamics constrains the ratio between the forward and backward values, not the ratio of values.


Non-Michaelis–Menten kinetics
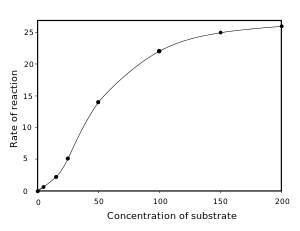
Many different enzyme systems follow non Michaelis-Menten behavior. A select few examples include kinetics of self-catalytic enzymes, cooperative and allosteric enzymes, interfacial and intracellular enzymes, processive enzymes and so forth. Some enzymes produce a sigmoid v by [S] plot, which often indicates cooperative binding of substrate to the active site. This means that the binding of one substrate molecule affects the binding of subsequent substrate molecules. This behavior is most common in multimeric enzymes with several interacting active sites.[32] Here, the mechanism of cooperation is similar to that of hemoglobin, with binding of substrate to one active site altering the affinity of the other active sites for substrate molecules. Positive cooperativity occurs when binding of the first substrate molecule increases the affinity of the other active sites for substrate. Negative cooperativity occurs when binding of the first substrate decreases the affinity of the enzyme for other substrate molecules.
Allosteric enzymes include mammalian tyrosyl tRNA-synthetase, which shows negative cooperativity,[33] and bacterial aspartate transcarbamoylase[34] and phosphofructokinase,[35] which show positive cooperativity.
Cooperativity is surprisingly common and can help regulate the responses of enzymes to changes in the concentrations of their substrates. Positive cooperativity makes enzymes much more sensitive to [S] and their activities can show large changes over a narrow range of substrate concentration. Conversely, negative cooperativity makes enzymes insensitive to small changes in [S].
The Hill equation[36] is often used to describe the degree of cooperativity quantitatively in non-Michaelis–Menten kinetics. The derived Hill coefficient n measures how much the binding of substrate to one active site affects the binding of substrate to the other active sites. A Hill coefficient of <1 indicates negative cooperativity and a coefficient of >1 indicates positive cooperativity.
Pre-steady-state kinetics

In the first moment after an enzyme is mixed with substrate, no product has been formed and no intermediates exist. The study of the next few milliseconds of the reaction is called pre-steady-state kinetics. Pre-steady-state kinetics is therefore concerned with the formation and consumption of enzyme–substrate intermediates (such as ES or E*) until their steady-state concentrations are reached.
This approach was first applied to the hydrolysis reaction catalysed by chymotrypsin.[37] Often, the detection of an intermediate is a vital piece of evidence in investigations of what mechanism an enzyme follows. For example, in the ping–pong mechanisms that are shown above, rapid kinetic measurements can follow the release of product P and measure the formation of the modified enzyme intermediate E*.[38] In the case of chymotrypsin, this intermediate is formed by an attack on the substrate by the nucleophilic serine in the active site and the formation of the acyl-enzyme intermediate.
In the figure to the right, the enzyme produces E* rapidly in the first few seconds of the reaction. The rate then slows as steady state is reached. This rapid burst phase of the reaction measures a single turnover of the enzyme. Consequently, the amount of product released in this burst, shown as the intercept on the y-axis of the graph, also gives the amount of functional enzyme which is present in the assay.[39]
Chemical mechanism
An important goal of measuring enzyme kinetics is to determine the chemical mechanism of an enzyme reaction, i.e., the sequence of chemical steps that transform substrate into product. The kinetic approaches discussed above will show at what rates intermediates are formed and inter-converted, but they cannot identify exactly what these intermediates are.
Kinetic measurements taken under various solution conditions or on slightly modified enzymes or substrates often shed light on this chemical mechanism, as they reveal the rate-determining step or intermediates in the reaction. For example, the breaking of a covalent bond to a hydrogen atom is a common rate-determining step. Which of the possible hydrogen transfers is rate determining can be shown by measuring the kinetic effects of substituting each hydrogen by deuterium, its stable isotope. The rate will change when the critical hydrogen is replaced, due to a primary kinetic isotope effect, which occurs because bonds to deuterium are harder to break than bonds to hydrogen.[40] It is also possible to measure similar effects with other isotope substitutions, such as 13C/12C and 18O/16O, but these effects are more subtle.[41]
Isotopes can also be used to reveal the fate of various parts of the substrate molecules in the final products. For example, it is sometimes difficult to discern the origin of an oxygen atom in the final product; since it may have come from water or from part of the substrate. This may be determined by systematically substituting oxygen's stable isotope 18O into the various molecules that participate in the reaction and checking for the isotope in the product.[42] The chemical mechanism can also be elucidated by examining the kinetics and isotope effects under different pH conditions,[43] by altering the metal ions or other bound cofactors,[44] by site-directed mutagenesis of conserved amino acid residues, or by studying the behaviour of the enzyme in the presence of analogues of the substrate(s).[45]
Enzyme inhibition and activation
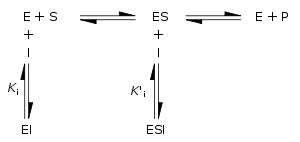
Enzyme inhibitors are molecules that reduce or abolish enzyme activity, while enzyme activators are molecules that increase the catalytic rate of enzymes. These interactions can be either reversible (i.e., removal of the inhibitor restores enzyme activity) or irreversible (i.e., the inhibitor permanently inactivates the enzyme).
Reversible inhibitors
Traditionally reversible enzyme inhibitors have been classified as competitive, uncompetitive, or non-competitive, according to their effects on KM and Vmax. These different effects result from the inhibitor binding to the enzyme E, to the enzyme–substrate complex ES, or to both, respectively. The division of these classes arises from a problem in their derivation and results in the need to use two different binding constants for one binding event. The binding of an inhibitor and its effect on the enzymatic activity are two distinctly different things, another problem the traditional equations fail to acknowledge. In noncompetitive inhibition the binding of the inhibitor results in 100% inhibition of the enzyme only, and fails to consider the possibility of anything in between.[46] In noncompetitive inhibition, the inhibitor will bind to an enzyme at its allosteric site; therefore, the binding affinity, or inverse of KM, of the substrate with the enzyme will remain the same. On the other hand, the Vmax will decrease relative to an uninhibited enzyme. On a Lineweaver-Burk plot, the presence of a noncompetitive inhibitor is illustrated by a change in the y-intercept, defined as 1/Vmax. The x-intercept, defined as −1/KM, will remain the same. In competitive inhibition, the inhibitor will bind to an enzyme at the active site, competing with the substrate. As a result, the KM will increase and the Vmax will remain the same.[47] The common form of the inhibitory term also obscures the relationship between the inhibitor binding to the enzyme and its relationship to any other binding term be it the Michaelis–Menten equation or a dose response curve associated with ligand receptor binding. To demonstrate the relationship the following rearrangement can be made:
![{\displaystyle {\cfrac {V_{\max }}{1+{\cfrac {[I]}{K_{i}}}}}={\cfrac {V_{\max }}{\cfrac {[I]+K_{i}}{K_{i}}}}}](../I/1c58cef49731511f5011822f1e92d4da22814891.svg)
Adding zero to the bottom ([I]-[I])
![{\cfrac {V_{\max }}{\cfrac {[I]+K_{i}}{[I]+K_{i}-[I]}}}](../I/42e34b0927bf9484b8a664e022d3fd6ba0ad2326.svg)
Dividing by [I]+Ki
![{\displaystyle {\cfrac {V_{\max }}{\cfrac {1}{1-{\cfrac {[I]}{[I]+K_{i}}}}}}=V_{\max }-V_{\max }{\cfrac {[I]}{[I]+K_{i}}}}](../I/4132fbd61b7474f8e3ef391f82d1d196a2a325ff.svg)
This notation demonstrates that similar to the Michaelis–Menten equation, where the rate of reaction depends on the percent of the enzyme population interacting with substrate, the effect of the inhibitor is a result of the percent of the enzyme population interacting with inhibitor. The only problem with this equation in its present form is that it assumes absolute inhibition of the enzyme with inhibitor binding, when in fact there can be a wide range of effects anywhere from 100% inhibition of substrate turn over to just >0%. To account for this the equation can be easily modified to allow for different degrees of inhibition by including a delta Vmax term.
![V_{\max }-\Delta V_{\max }{\cfrac {[I]}{[I]+K_{i}}}](../I/90f5601fefd8114c165ac3dfb739e0642e62610c.svg)
or
![{\displaystyle V_{\max 1}-(V_{\max 1}-V_{\max 2}){\cfrac {[I]}{[I]+K_{i}}}}](../I/8c31b5f56aaeed122d1f8a67491c56d272686b6e.svg)
This term can then define the residual enzymatic activity present when the inhibitor is interacting with individual enzymes in the population. However the inclusion of this term has the added value of allowing for the possibility of activation if the secondary Vmax term turns out to be higher than the initial term. To account for the possibly of activation as well the notation can then be rewritten replacing the inhibitor "I" with a modifier term denoted here as "X".
![{\displaystyle V_{\max 1}-(V_{\max 1}-V_{\max 2}){\cfrac {[X]}{[X]+K_{x}}}}](../I/2bb687fca77a00dba879a00fb32b7ca1bc867973.svg)
While this terminology results in a simplified way of dealing with kinetic effects relating to the maximum velocity of the Michaelis–Menten equation, it highlights potential problems with the term used to describe effects relating to the KM. The KM relating to the affinity of the enzyme for the substrate should in most cases relate to potential changes in the binding site of the enzyme which would directly result from enzyme inhibitor interactions. As such a term similar to the one proposed above to modulate Vmax should be appropriate in most situations:[48]
![{\displaystyle K_{m1}-(K_{m1}-K_{m2}){\cfrac {[X]}{[X]+K_{x}}}}](../I/55a298dbb441e88b96a618cc31df3aaebac17d85.svg)
Irreversible inhibitors
Enzyme inhibitors can also irreversibly inactivate enzymes, usually by covalently modifying active site residues. These reactions, which may be called suicide substrates, follow exponential decay functions and are usually saturable. Below saturation, they follow first order kinetics with respect to inhibitor. Irreversible inhibition could be classified into two distinct types. Affinity labelling is a type of irreversible inhibition where a functional group that is highly reactive modifies a catalytically critical residue on the protein of interest to bring about inhibition. Mechanism-based inhibition, on the other hand, involves binding of the inhibitor followed by enzyme mediated alterations that transform the latter into a reactive group that irreversibly modifies the enzyme.
Philosophical discourse on reversibility and irreversibility of inhibition
Having discussed reversible inhibition and irreversible inhibition in the above two headings, it would have to be pointed out that the concept of reversibility (or irreversibility) is a purely theoretical construct exclusively dependent on the time-frame of the assay, i.e., a reversible assay involving association and dissociation of the inhibitor molecule in the minute timescales would seem irreversible if an assay assess the outcome in the seconds and vice versa. There is a continuum of inhibitor behaviors spanning reversibility and irreversibility at a given non-arbitrary assay time frame. There are inhibitors that show slow-onset behavior and most of these inhibitors, invariably, also show tight-binding to the protein target of interest.
Mechanisms of catalysis

The favoured model for the enzyme–substrate interaction is the induced fit model.[49] This model proposes that the initial interaction between enzyme and substrate is relatively weak, but that these weak interactions rapidly induce conformational changes in the enzyme that strengthen binding. These conformational changes also bring catalytic residues in the active site close to the chemical bonds in the substrate that will be altered in the reaction.[50] Conformational changes can be measured using circular dichroism or dual polarisation interferometry. After binding takes place, one or more mechanisms of catalysis lower the energy of the reaction's transition state by providing an alternative chemical pathway for the reaction. Mechanisms of catalysis include catalysis by bond strain; by proximity and orientation; by active-site proton donors or acceptors; covalent catalysis and quantum tunnelling.[38][51]
Enzyme kinetics cannot prove which modes of catalysis are used by an enzyme. However, some kinetic data can suggest possibilities to be examined by other techniques. For example, a ping–pong mechanism with burst-phase pre-steady-state kinetics would suggest covalent catalysis might be important in this enzyme's mechanism. Alternatively, the observation of a strong pH effect on Vmax but not KM might indicate that a residue in the active site needs to be in a particular ionisation state for catalysis to occur.
History
In 1902 Victor Henri proposed a quantitative theory of enzyme kinetics,[52] but at the time the experimental significance of the hydrogen ion concentration was not yet recognized. After Peter Lauritz Sørensen had defined the logarithmic pH-scale and introduced the concept of buffering in 1909[53] the German chemist Leonor Michaelis and Dr. Maud Leonora Menten (a postdoctoral researcher in Michaelis's lab at the time) repeated Henri's experiments and confirmed his equation, which is now generally referred to as Michaelis-Menten kinetics (sometimes also Henri-Michaelis-Menten kinetics).[54] Their work was further developed by G. E. Briggs and J. B. S. Haldane, who derived kinetic equations that are still widely considered today a starting point in modeling enzymatic activity.[55]
The major contribution of the Henri-Michaelis-Menten approach was to think of enzyme reactions in two stages. In the first, the substrate binds reversibly to the enzyme, forming the enzyme-substrate complex. This is sometimes called the Michaelis complex. The enzyme then catalyzes the chemical step in the reaction and releases the product. The kinetics of many enzymes is adequately described by the simple Michaelis-Menten model, but all enzymes have internal motions that are not accounted for in the model and can have significant contributions to the overall reaction kinetics. This can be modeled by introducing several Michaelis-Menten pathways that are connected with fluctuating rates,[56][57][58] which is a mathematical extension of the basic Michaelis Menten mechanism.[59]
Software
ENZO (Enzyme Kinetics) is a graphical interface tool for building kinetic models of enzyme catalyzed reactions. ENZO automatically generates the corresponding differential equations from a stipulated enzyme reaction scheme. These differential equations are processed by a numerical solver and a regression algorithm which fits the coefficients of differential equations to experimentally observed time course curves. ENZO allows rapid evaluation of rival reaction schemes and can be used for routine tests in enzyme kinetics.[60]
See also
Footnotes
References
- ↑ Wrighton MS, Ebbing DD (1993). General chemistry (4th ed.). Boston: Houghton Mifflin. ISBN 978-0-395-63696-1.
- 1 2 Fromm H.J., Hargrove M.S. (2012) Enzyme Kinetics. In: Essentials of Biochemistry. Springer, Berlin, Heidelberg
- ↑ Danson M, Eisenthal R (2002). Enzyme assays: a practical approach. Oxford [Oxfordshire]: Oxford University Press. ISBN 978-0-19-963820-8.
- ↑ Xie XS, Lu HP (June 1999). "Single-molecule enzymology". The Journal of Biological Chemistry. 274 (23): 15967–15970. doi:10.1074/jbc.274.23.15967. PMID 10347141.
- ↑ Lu HP (June 2004). "Single-molecule spectroscopy studies of conformational change dynamics in enzymatic reactions". Current Pharmaceutical Biotechnology. 5 (3): 261–269. doi:10.2174/1389201043376887. PMID 15180547.
- ↑ Schnell JR, Dyson HJ, Wright PE (2004). "Structure, dynamics, and catalytic function of dihydrofolate reductase". Annual Review of Biophysics and Biomolecular Structure. 33: 119–140. doi:10.1146/annurev.biophys.33.110502.133613. PMID 15139807.
- ↑ Gibson QH (1969). "[6] Rapid mixing: Stopped flow". Rapid mixing: Stopped flow. Methods in Enzymology. Vol. 16. pp. 187–228. doi:10.1016/S0076-6879(69)16009-7. ISBN 978-0-12-181873-9.
- ↑ Duggleby RG (1995). "[3] Analysis of enzyme progress curves by nonlinear regression". Analysis of enzyme progress curves by non-linear regression. Methods in Enzymology. Vol. 249. pp. 61–90. doi:10.1016/0076-6879(95)49031-0. ISBN 978-0-12-182150-0. PMID 7791628.
- ↑ Murray JB, Dunham CM, Scott WG (January 2002). "A pH-dependent conformational change, rather than the chemical step, appears to be rate-limiting in the hammerhead ribozyme cleavage reaction". Journal of Molecular Biology. 315 (2): 121–130. doi:10.1006/jmbi.2001.5145. PMID 11779233. S2CID 18102624.
- ↑ Michaelis L. and Menten M.L. Kinetik der Invertinwirkung Biochem. Z. 1913; 49:333–369 English translation Accessed 6 April 2007
- ↑ Stroppolo ME, Falconi M, Caccuri AM, Desideri A (September 2001). "Superefficient enzymes". Cellular and Molecular Life Sciences. 58 (10): 1451–1460. doi:10.1007/PL00000788. PMID 11693526. S2CID 24874575.
- ↑ Bar-Even A, Noor E, Savir Y, Liebermeister W, Davidi D, Tawfik DS, Milo R (May 2011). "The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters". Biochemistry. 50 (21): 4402–4410. doi:10.1021/bi2002289. PMID 21506553.
- ↑ Walsh R, Martin E, Darvesh S (January 2010). "A method to describe enzyme-catalyzed reactions by combining steady state and time course enzyme kinetic parameters". Biochimica et Biophysica Acta (BBA) - General Subjects. 1800 (1): 1–5. doi:10.1016/j.bbagen.2009.10.007. PMID 19840832.
- ↑ Beal SL (December 1983). "Computation of the explicit solution to the Michaelis-Menten equation". Journal of Pharmacokinetics and Biopharmaceutics. 11 (6): 641–657. doi:10.1007/BF01059062. PMID 6689584. S2CID 32571415.
- ↑ Schnell S, Mendoza C (1997). "Closed Form Solution for Time-dependent Enzyme Kinetics". Journal of Theoretical Biology. 187 (2): 207–212. Bibcode:1997JThBi.187..207S. doi:10.1006/jtbi.1997.0425.
- ↑ Goudar CT, Sonnad JR, Duggleby RG (January 1999). "Parameter estimation using a direct solution of the integrated Michaelis-Menten equation" (PDF). Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology. 1429 (2): 377–383. doi:10.1016/s0167-4838(98)00247-7. PMID 9989222. Archived from the original (PDF) on 9 November 2015.
- ↑ Goudar CT, Harris SK, McInerney MJ, Suflita JM (December 2004). "Progress curve analysis for enzyme and microbial kinetic reactions using explicit solutions based on the Lambert W function". Journal of Microbiological Methods. 59 (3): 317–326. doi:10.1016/j.mimet.2004.06.013. PMID 15488275.
- ↑ Berberan-Santos MN (2010). "A General Treatment of Henri Michaelis Menten Enzyme Kinetics: Exact Series Solution and Approximate Analytical Solutions" (PDF). MATCH Communications in Mathematical and in Computer Chemistry. 63: 283.
- ↑ Jones ME (December 1992). "Analysis of algebraic weighted least-squares estimators for enzyme parameters". The Biochemical Journal. 288 ( Pt 2) (Pt 2): 533–538. doi:10.1042/bj2880533. PMC 1132043. PMID 1463456.
- ↑ Tseng SJ, Hsu JP (August 1990). "A comparison of the parameter estimating procedures for the Michaelis-Menten model". Journal of Theoretical Biology. 145 (4): 457–464. Bibcode:1990JThBi.145..457T. doi:10.1016/S0022-5193(05)80481-3. PMID 2246896.
- ↑ Bravo IG, Busto F, De Arriaga D, Ferrero MA, Rodríguez-Aparicio LB, Martínez-Blanco H, Reglero A (September 2001). "A normalized plot as a novel and time-saving tool in complex enzyme kinetic analysis". The Biochemical Journal. 358 (Pt 3): 573–583. doi:10.1042/bj3580573. PMC 1222113. PMID 11577687.
- ↑ Almaas E, Kovács B, Vicsek T, Oltvai ZN, Barabási AL (February 2004). "Global organization of metabolic fluxes in the bacterium Escherichia coli". Nature. 427 (6977): 839–843. arXiv:q-bio/0403001. Bibcode:2004Natur.427..839A. doi:10.1038/nature02289. PMID 14985762. S2CID 715721.
- ↑ Reed JL, Vo TD, Schilling CH, Palsson BO (2003). "An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR)". Genome Biology. 4 (9): R54. doi:10.1186/gb-2003-4-9-r54. PMC 193654. PMID 12952533.
- ↑ for a complete derivation, see here
- ↑ Dirr H, Reinemer P, Huber R (March 1994). "X-ray crystal structures of cytosolic glutathione S-transferases. Implications for protein architecture, substrate recognition and catalytic function". European Journal of Biochemistry. 220 (3): 645–661. doi:10.1111/j.1432-1033.1994.tb18666.x. PMID 8143720.
- ↑ Stone SR, Morrison JF (July 1988). "Dihydrofolate reductase from Escherichia coli: the kinetic mechanism with NADPH and reduced acetylpyridine adenine dinucleotide phosphate as substrates". Biochemistry. 27 (15): 5493–5499. doi:10.1021/bi00415a016. PMID 3052577.
- ↑ Fisher PA (1994). Enzymologic mechanism of replicative DNA polymerases in higher eukaryotes. Progress in Nucleic Acid Research and Molecular Biology. Vol. 47. pp. 371–97. doi:10.1016/S0079-6603(08)60257-3. ISBN 978-0-12-540047-3. PMID 8016325.
- ↑ Akerman SE, Müller S (August 2003). "2-Cys peroxiredoxin PfTrx-Px1 is involved in the antioxidant defence of Plasmodium falciparum". Molecular and Biochemical Parasitology. 130 (2): 75–81. doi:10.1016/S0166-6851(03)00161-0. PMID 12946843.
- ↑ Bravo IG, Barrallo S, Ferrero MA, Rodríguez-Aparicio LB, Martínez-Blanco H, Reglero A (September 2001). "Kinetic properties of the acylneuraminate cytidylyltransferase from Pasteurella haemolytica A2". The Biochemical Journal. 358 (Pt 3): 585–598. doi:10.1042/bj3580585. PMC 1222114. PMID 11577688.
- ↑ Kraut J (1977). "Serine proteases: structure and mechanism of catalysis". Annual Review of Biochemistry. 46: 331–358. doi:10.1146/annurev.bi.46.070177.001555. PMID 332063.
- ↑ Cornish-Bowden A (2012). Fundamentals of Enzyme Kinetics. Wiley-Blackwell.
Some enzymes are much more effective catalysts for one direction than the other. As a striking example, the limiting rates of the forward reaction catalyzed by methionine adenosyltransferase is about 105 greater than that for the reverse direction, even though the equilibrium constant is close to unity (page 59).
- ↑ Ricard J, Cornish-Bowden A (July 1987). "Co-operative and allosteric enzymes: 20 years on". European Journal of Biochemistry. 166 (2): 255–272. doi:10.1111/j.1432-1033.1987.tb13510.x. PMID 3301336.
- ↑ Ward WH, Fersht AR (July 1988). "Tyrosyl-tRNA synthetase acts as an asymmetric dimer in charging tRNA. A rationale for half-of-the-sites activity". Biochemistry. 27 (15): 5525–5530. doi:10.1021/bi00415a021. PMID 3179266.
- ↑ Helmstaedt K, Krappmann S, Braus GH (September 2001). "Allosteric regulation of catalytic activity: Escherichia coli aspartate transcarbamoylase versus yeast chorismate mutase". Microbiology and Molecular Biology Reviews. 65 (3): 404–21, table of contents. doi:10.1128/MMBR.65.3.404-421.2001. PMC 99034. PMID 11528003.
- ↑ Schirmer T, Evans PR (January 1990). "Structural basis of the allosteric behaviour of phosphofructokinase". Nature. 343 (6254): 140–145. Bibcode:1990Natur.343..140S. doi:10.1038/343140a0. PMID 2136935. S2CID 4272821.
- ↑ Hill AV (1910). "The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves". J. Physiol. 40: iv–vii.
- ↑ Hartley BS, Kilby BA (February 1954). "The reaction of p-nitrophenyl esters with chymotrypsin and insulin". The Biochemical Journal. 56 (2): 288–297. doi:10.1042/bj0560288. PMC 1269615. PMID 13140189.
- 1 2 Fersht, Alan (1999). Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. San Francisco: W.H. Freeman. ISBN 978-0-7167-3268-6.
- ↑ Bender ML, Begué-Cantón ML, Blakeley RL, Brubacher LJ, Feder J, Gunter CR, et al. (December 1966). "The determination of the concentration of hydrolytic enzyme solutions: alpha-chymotrypsin, trypsin, papain, elastase, subtilisin, and acetylcholinesterase". Journal of the American Chemical Society. 88 (24): 5890–5913. doi:10.1021/ja00976a034. PMID 5980876.
- ↑ Cleland WW (January 2005). "The use of isotope effects to determine enzyme mechanisms". Archives of Biochemistry and Biophysics. 433 (1): 2–12. doi:10.1016/j.abb.2004.08.027. PMID 15581561.
- ↑ Northrop DB (1981). "The expression of isotope effects on enzyme-catalyzed reactions". Annual Review of Biochemistry. 50: 103–131. doi:10.1146/annurev.bi.50.070181.000535. PMID 7023356.
- ↑ Baillie TA, Rettenmeier AW (1986). "Drug biotransformation: mechanistic studies with stable isotopes". Journal of Clinical Pharmacology. 26 (6): 448–451. doi:10.1002/j.1552-4604.1986.tb03556.x. PMID 3734135. S2CID 39193680.
- ↑ Cleland WW (1982). "Use of isotope effects to elucidate enzyme mechanisms". CRC Critical Reviews in Biochemistry. 13 (4): 385–428. doi:10.3109/10409238209108715. PMID 6759038.
- ↑ Christianson DW, Cox JD (1999). "Catalysis by metal-activated hydroxide in zinc and manganese metalloenzymes". Annual Review of Biochemistry. 68: 33–57. doi:10.1146/annurev.biochem.68.1.33. PMID 10872443.
- ↑ Kraut DA, Carroll KS, Herschlag D (2003). "Challenges in enzyme mechanism and energetics". Annual Review of Biochemistry. 72: 517–571. doi:10.1146/annurev.biochem.72.121801.161617. PMID 12704087.
- ↑ Walsh R, Martin E, Darvesh S (December 2011). "Limitations of conventional inhibitor classifications". Integrative Biology. 3 (12): 1197–1201. doi:10.1039/c1ib00053e. PMID 22038120.
- ↑ Cleland WW (February 1963). "The kinetics of enzyme-catalyzed reactions with two or more substrates or products. III. Prediction of initial velocity and inhibition patterns by inspection". Biochimica et Biophysica Acta. 67: 188–196. doi:10.1016/0006-3002(63)91816-x. PMID 14021669.
- ↑ Walsh R, Martin E, Darvesh S (May 2007). "A versatile equation to describe reversible enzyme inhibition and activation kinetics: modeling beta-galactosidase and butyrylcholinesterase". Biochimica et Biophysica Acta (BBA) - General Subjects. 1770 (5): 733–746. doi:10.1016/j.bbagen.2007.01.001. PMID 17307293.
- ↑ Koshland DE (February 1958). "Application of a Theory of Enzyme Specificity to Protein Synthesis". Proceedings of the National Academy of Sciences of the United States of America. 44 (2): 98–104. Bibcode:1958PNAS...44...98K. doi:10.1073/pnas.44.2.98. PMC 335371. PMID 16590179.
- ↑ Hammes GG (July 2002). "Multiple conformational changes in enzyme catalysis". Biochemistry. 41 (26): 8221–8228. doi:10.1021/bi0260839. PMID 12081470.
- ↑ Sutcliffe MJ, Scrutton NS (July 2002). "A new conceptual framework for enzyme catalysis. Hydrogen tunnelling coupled to enzyme dynamics in flavoprotein and quinoprotein enzymes". European Journal of Biochemistry. 269 (13): 3096–3102. doi:10.1046/j.1432-1033.2002.03020.x. PMID 12084049.
- ↑ Henri V (1902). "Theorie generale de l'action de quelques diastases". Compt. Rend. Acad. Sci. Paris. 135: 916–9.
- ↑ Sørensen PL (1909). "Enzymstudien {II}. Über die Messung und Bedeutung der Wasserstoffionenkonzentration bei enzymatischen Prozessen" [Enzyme studies III: About the measurement and significance of the hydrogen ion concentration in enzymatic processes]. Biochem. Z. (in German). 21: 131–304.
- ↑ Michaelis L, Menten M (1913). "Die Kinetik der Invertinwirkung" [The Kinetics of Invertase Action]. Biochem. Z. (in German). 49: 333–369.; Michaelis L, Menten ML, Johnson KA, Goody RS (October 2011). "The original Michaelis constant: translation of the 1913 Michaelis-Menten paper". Biochemistry. 50 (39): 8264–8269. doi:10.1021/bi201284u. PMC 3381512. PMID 21888353.
- ↑ Briggs GE, Haldane JB (1925). "A Note on the Kinetics of Enzyme Action". The Biochemical Journal. 19 (2): 338–339. doi:10.1042/bj0190338. PMC 1259181. PMID 16743508.
- ↑ Flomenbom O, Velonia K, Loos D, Masuo S, Cotlet M, Engelborghs Y, et al. (February 2005). "Stretched exponential decay and correlations in the catalytic activity of fluctuating single lipase molecules". Proceedings of the National Academy of Sciences of the United States of America. 102 (7): 2368–2372. Bibcode:2005PNAS..102.2368F. doi:10.1073/pnas.0409039102. PMC 548972. PMID 15695587.
- ↑ English BP, Min W, van Oijen AM, Lee KT, Luo G, Sun H, et al. (February 2006). "Ever-fluctuating single enzyme molecules: Michaelis-Menten equation revisited". Nature Chemical Biology. 2 (2): 87–94. doi:10.1038/nchembio759. PMID 16415859. S2CID 2201882.
- ↑ Lu HP, Xun L, Xie XS (December 1998). "Single-molecule enzymatic dynamics". Science. 282 (5395): 1877–1882. Bibcode:1998Sci...282.1877P. doi:10.1126/science.282.5395.1877. PMID 9836635.
- ↑ Xue X, Liu F, Ou-Yang ZC (September 2006). "Single molecule Michaelis-Menten equation beyond quasistatic disorder". Physical Review E. 74 (3 Pt 1): 030902. arXiv:cond-mat/0604364. Bibcode:2006PhRvE..74c0902X. doi:10.1103/PhysRevE.74.030902. PMID 17025584. S2CID 41674948.
- ↑ Bevc S, Konc J, Stojan J, Hodošček M, Penca M, Praprotnik M, Janežič D (2011). "ENZO: a web tool for derivation and evaluation of kinetic models of enzyme catalyzed reactions". PLOS ONE. 6 (7): e22265. Bibcode:2011PLoSO...622265B. doi:10.1371/journal.pone.0022265. PMC 3139599. PMID 21818304. ENZO server
Further reading
Introductory
- Cornish-Bowden A (2012). Fundamentals of enzyme kinetics (4th ed.). Weinheim: Wiley-Blackwell. ISBN 978-3-527-33074-4.
- Stevens L, Price NC (1999). Fundamentals of enzymology: the cell and molecular biology of catalytic proteins. Oxford [Oxfordshire]: Oxford University Press. ISBN 978-0-19-850229-6.
- Bugg T (2004). Introduction to Enzyme and Coenzyme Chemistry. Cambridge, MA: Blackwell Publishers. ISBN 978-1-4051-1452-3.
- Segel IH (1993). Enzyme kinetics: behavior and analysis of rapid equilibrium and steady state enzyme systems. New York: Wiley. ISBN 978-0-471-30309-1.
Advanced
- Fersht A (1999). Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. San Francisco: W.H. Freeman. ISBN 978-0-7167-3268-6.
- Schnell S, Maini PK (2004). "A century of enzyme kinetics: Reliability of the KM and vmax estimates". Comments on Theoretical Biology. 8 (2–3): 169–87. CiteSeerX 10.1.1.493.7178. doi:10.1080/08948550302453.
- Walsh C (1979). Enzymatic reaction mechanisms. San Francisco: W. H. Freeman. ISBN 978-0-7167-0070-8.
- Cleland WW, Cook P (2007). Enzyme kinetics and mechanism. New York: Garland Science. ISBN 978-0-8153-4140-6.
External links
- Animation of an enzyme assay — Shows effects of manipulating assay conditions
- MACiE — A database of enzyme reaction mechanisms
- ENZYME — Expasy enzyme nomenclature database
- ENZO — Web application for easy construction and quick testing of kinetic models of enzyme catalyzed reactions.
- ExCatDB — A database of enzyme catalytic mechanisms
- BRENDA — Comprehensive enzyme database, giving substrates, inhibitors and reaction diagrams
- SABIO-RK — A database of reaction kinetics
- Joseph Kraut's Research Group, University of California San Diego — Animations of several enzyme reaction mechanisms
- Symbolism and Terminology in Enzyme Kinetics — A comprehensive explanation of concepts and terminology in enzyme kinetics
- An introduction to enzyme kinetics — An accessible set of on-line tutorials on enzyme kinetics
- Enzyme kinetics animated tutorial — An animated tutorial with audio