In organic chemistry, an electrocyclic reaction is a type of pericyclic rearrangement where the net result is one pi bond being converted into one sigma bond or vice versa.[1] These reactions are usually categorized by the following criteria:
- Reactions can be either photochemical or thermal.
- Reactions can be either ring-opening or ring-closing (electrocyclization).
- Depending on the type of reaction (photochemical or thermal) and the number of pi electrons, the reaction can happen through either a conrotatory or disrotatory mechanism.
- The type of rotation determines whether the cis or trans isomer of the product will be formed.
Classical examples
The Nazarov cyclization reaction is a named electrocyclic reaction converting divinylketones to cyclopentenones.
A classic example is the thermal ring-opening reaction of 3,4-dimethylcyclobutene. The cis isomer exclusively yields cis,trans-hexa-2,4-diene whereas the trans isomer gives the trans,trans diene:[2]
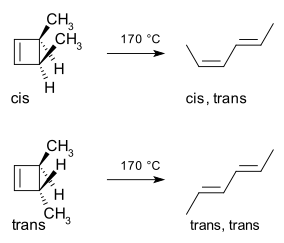
This reaction course can be explained in a simple analysis through the frontier-orbital method: the sigma bond in the reactant will open in such a way that the resulting p-orbitals will have the same symmetry as the HOMO of the product (a hexadiene). The only way to accomplish this is through a conrotatory ring-opening which results in opposite phases for the terminal lobes.

Stereospecificity of electrocyclic reactions
When performing an electrocyclic reaction, it is often desirable to predict the cis/trans geometry of the reaction's product. The first step in this process is to determine whether a reaction proceeds through conrotation or disrotation. The table below shows the selectivity rules for thermal and photochemical electrocyclic reactions.
| System | Thermally induced (ground state) | Photochemically induced (excited state) |
|---|---|---|
| Even # of conjugation | Conrotatory | Disrotatory |
| Odd # of conjugation | Disrotatory | Conrotatory |
For the example given below, the thermal reaction of (trans,cis,trans)-octa-2,4,6-triene will happen through a disrotatory mechanism. After determining the type of rotation, whether the product will be cis or trans can be determined by examining the starting molecule. In the example below, the disrotation causes both methyls to point upwards, causing the product to be cis-dimethylcyclohexadiene.
In addition, the torquoselectivity in an electrocyclic reaction refers to the direction of rotation. For example, a reaction that is conrotatory can still rotate in two directions, producing enantiomeric products. A reaction that is torquoselective restricts one of these directions of rotation (partially or completely) to produce a product in enantiomeric excess.

Mechanism of thermal reactions
Woodward–Hoffmann rules

Correlation diagrams, which connect the molecular orbitals of the reactant to those of the product having the same symmetry, can then be constructed for the two processes.[3]
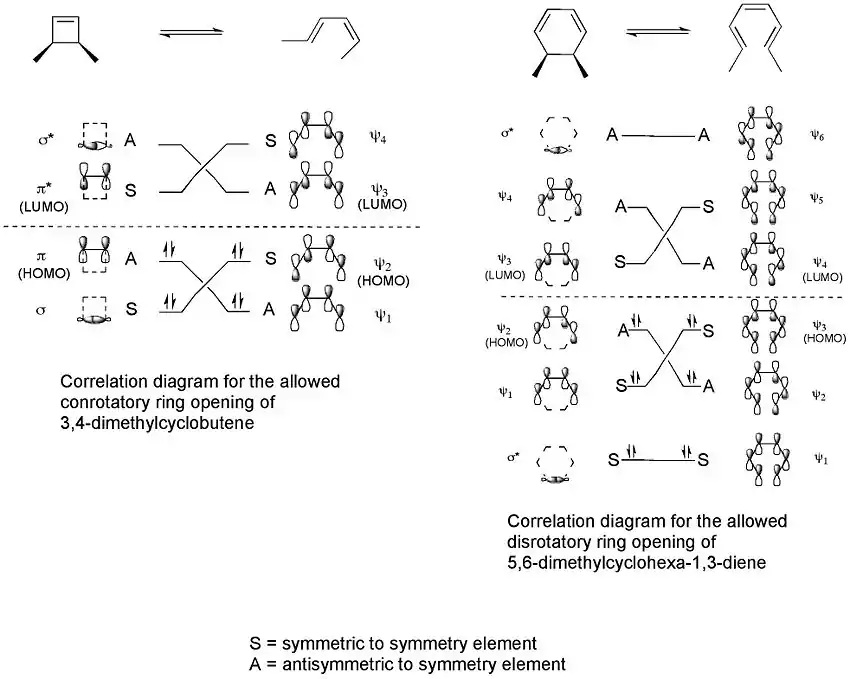
These correlation diagrams indicate that only a conrotatory ring opening of 3,4-dimethylcyclobutene is symmetry allowed whereas only a disrotatory ring opening of 5,6-dimethylcyclohexa-1,3-diene is symmetry allowed. This is because only in these cases would maximum orbital overlap occur in the transition state. Also, the formed product would be in a ground state rather than an excited state.
Frontier molecular orbital theory
According to the frontier molecular orbital theory, the sigma bond in the ring will open in such a way that the resulting p-orbitals will have the same symmetry as the HOMO of the product.[4]

For the 5,6-dimethylcyclohexa-1,3-diene, only a disrotatory mode would result in p-orbitals having the same symmetry as the HOMO of hexatriene. For the 3,4-dimethylcyclobutene, on the other hand, only a conrotatory mode would result in p-orbitals having the same symmetry as the HOMO of butadiene.
Mechanism of photochemical reactions
If the ring opening of 3,4-dimethylcyclobutene were carried out under photochemical conditions the resulting electrocyclization would be occur through a disrotatory mode instead of a conrotatory mode as can be seen by the correlation diagram for the allowed excited state ring opening reaction.

Only a disrotatory mode, in which symmetry about a reflection plane is maintained throughout the reaction, would result in maximum orbital overlap in the transition state. Also, once again, this would result in the formation of a product that is in an excited state of comparable stability to the excited state of the reactant compound.
Electrocyclic reactions in biological systems
Electrocyclic reactions occur frequently in nature.[5] One of the most common such electrocyclizations is the biosynthesis of vitamin D3.

The first step involves a photochemically induced conrotatory ring opening of 7-dehydrocholesterol to form pre vitamin D3. A [1,7]-hydride shift then forms vitamin D3.
Another example is in the proposed biosynthesis of aranotin, a naturally occurring oxepine, and its related compounds.

Enzymatic epoxidation of phenylalanine-derived diketopiperazine forms the arene oxide, which undergoes a 6π disrotatory ring opening electrocyclization reaction to produce the uncyclized oxepine. After a second epoxidation of the ring, the nearby nucleophilic nitrogen attacks the electrophilic carbon, forming a five membered ring. The resulting ring system is a common ring system found in aranotin and its related compounds.
The benzonorcaradiene diterpenoid (A) was rearranged into the benzocycloheptatriene diterpenoid isosalvipuberlin (B) by boiling a methylene chloride solution. This transformation can be envisaged as a disrotatory electrocyclic reaction, followed by two suprafacial 1,5-sigmatropic hydrogen shifts, as shown below.[6]

Electrocyclic reactions in organic synthesis
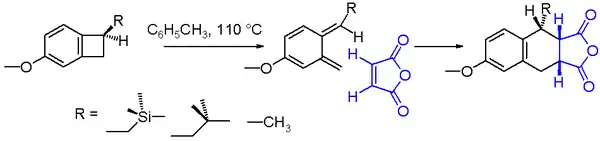
An often studied electrocyclic reaction is the conrotatory thermal ring-opening of benzocyclobutene. The reaction product is a very unstable ortho-quinodimethane but this molecule can be trapped in an endo addition with a strong dienophile such as maleic anhydride to the Diels-Alder adduct. The chemical yield for the ring opening of the benzocyclobutane depicted in scheme 2 is found to depend on the nature of the substituent R.[7] With a reaction solvent such as toluene and a reaction temperature of 110 °C, the yield increases going from methyl to isobutylmethyl to (trimethylsilyl)methyl. The increased reaction rate for the trimethylsilyl compound can be explained by silicon hyperconjugation as the βC-Si bond weakens the cyclobutane C-C bond by donating electrons.
A biomimetic electrocyclic cascade reaction was discovered in relation to the isolation and synthesis of certain endiandric acids:[8][9]

Asymmetric electrocyclic reactions are an emerging field in contemporary organic synthesis. The most commonly studied reactions in this field are the 4π Staudinger β-lactam synthesis[10] and the 4π Nazarov reaction; asymmetric catalysis of both reactions have been controlled by use of a chiral auxiliary, and the Nazarov reaction has been performed catalytically using chiral Lewis acids, Brønsted acids and chiral amines.[11]
References
- ↑ IUPAC Gold Book
- ↑ The preparation and isomerization of - and -3,4-dimethylcyclobutene. Tetrahedron Letters, Volume 6, Issue 17, 1965, Pages 1207-1212 Rudolph Ernst K. Winter doi:10.1016/S0040-4039(01)83997-6
- ↑ The conservation of orbital symmetry. Acc. Chem. Res., Volume 1, Issue 1, 1968, Pages 17–22 Roald Hoffmann and Robert B. Woodward doi:10.1021/ar50001a003
- ↑ Fleming, Ian. Frontier Orbitals and Organic Chemical Reactions. 1976 (John Wiley & Sons, Ltd.) ISBN 0-471-01820-1
- ↑ Biosynthetic and Biomimetic Electrocyclizations. Chem. Rev., Volume 105, Issue 12, 2005, Pages 4757-4778 Christopher M. Beaudry, Jeremiah P. Malerich, and Dirk Trauner doi:10.1021/cr0406110
- ↑ J. T. Arnason, Rachel Mata, John T. Romeo. Phytochemistry of Medicinal Plant(2nd Edition).1995 (Springer) ISBN 0-306-45181-6, ISBN 978-0-306-45181-2
- ↑ Accelerated Electrocyclic Ring-Opening of Benzocyclobutenes under the Influence of a -Silicon Atom Yuji Matsuya, Noriko Ohsawa, and Hideo Nemoto J. Am. Chem. Soc.; 2006; 128(2) pp 412 - 413; (Communication) doi:10.1021/ja055505+
- ↑ The endiandric acid cascade. Electrocyclizations in organic synthesis. 4. Biomimetic approach to endiandric acids A-G. Total synthesis and thermal studies K. C. Nicolaou, N. A. Petasis, R. E. Zipkin J. Am. Chem. Soc., 1982, 104 (20), pp 5560–5562 doi:10.1021/ja00384a080
- ↑ Inspirations, Discoveries, and Future Perspectives in Total Synthesis K. C. Nicolaou J. Org. Chem., 2009 Article ASAP doi:10.1021/jo802351b
- ↑ "Staudinger Synthesis".
- ↑ Asymmetric electrocyclic reactions, S. Thompson, A. G. Coyne, P. C. Knipe and M. D. Smith, Chem. Soc. Rev., 2011, 40, pp 4217-4231 doi:10.1039/C1CS15022G