| Diels-Alder reaction | |
|---|---|
| Reaction type | Cycloaddition |
| Identifiers | |
| Organic Chemistry Portal | diels-alder-reaction |
| RSC ontology ID | RXNO:0000006 |
_red.svg.png.webp)
In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally-allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes.[1][2] Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials.[3][4] The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels-Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.[5]
Mechanism
The reaction is an example of a concerted pericyclic reaction.[6] It is believed to occur via a single, cyclic transition state,[7] with no intermediates generated during the course of the reaction. As such, the Diels–Alder reaction is governed by orbital symmetry considerations: it is classified as a [π4s + π2s] cycloaddition, indicating that it proceeds through the suprafacial/suprafacial interaction of a 4π electron system (the diene structure) with a 2π electron system (the dienophile structure), an interaction that leads to a transition state without an additional orbital symmetry-imposed energetic barrier and allows the Diels–Alder reaction to take place with relative ease.[8]
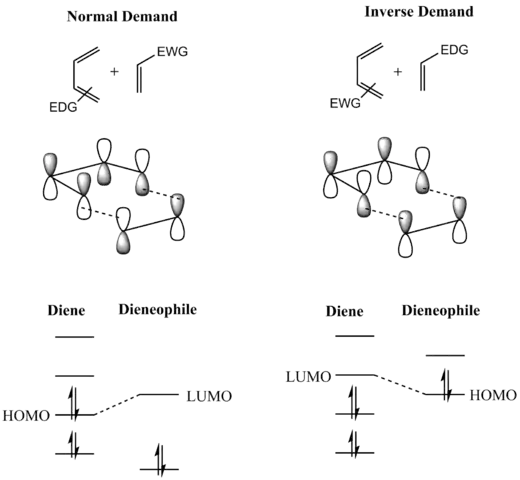
A consideration of the reactants' frontier molecular orbitals (FMO) makes plain why this is so. (The same conclusion can be drawn from an orbital correlation diagram or a Dewar-Zimmerman analysis.) For the more common "normal" electron demand Diels–Alder reaction, the more important of the two HOMO/LUMO interactions is that between the electron-rich diene's ψ2 as the highest occupied molecular orbital (HOMO) with the electron-deficient dienophile's π* as the lowest unoccupied molecular orbital (LUMO). However, the HOMO–LUMO energy gap is close enough that the roles can be reversed by switching electronic effects of the substituents on the two components. In an inverse (reverse) electron-demand Diels–Alder reaction, electron-withdrawing substituents on the diene lower the energy of its empty ψ3 orbital and electron-donating substituents on the dienophile raise the energy of its filled π orbital sufficiently that the interaction between these two orbitals becomes the most energetically significant stabilizing orbital interaction. Regardless of which situation pertains, the HOMO and LUMO of the components are in phase and a bonding interaction results as can be seen in the diagram below. Since the reactants are in their ground state, the reaction is initiated thermally and does not require activation by light.[8]
The "prevailing opinion"[9][10][11][12] is that most Diels–Alder reactions proceed through a concerted mechanism; the issue, however, has been thoroughly contested. Despite the fact that the vast majority of Diels–Alder reactions exhibit stereospecific, syn addition of the two components, a diradical intermediate has been postulated[7] (and supported with computational evidence) on the grounds that the observed stereospecificity does not rule out a two-step addition involving an intermediate that collapses to product faster than it can rotate to allow for inversion of stereochemistry.
There is a notable rate enhancement when certain Diels–Alder reactions are carried out in polar organic solvents such as dimethylformamide and ethylene glycol,[13] and even in water.[14] The reaction of cyclopentadiene and butenone for example is 700 times faster in water relative to 2,2,4-trimethylpentane as solvent.[14] Several explanations for this effect have been proposed, such as an increase in effective concentration due to hydrophobic packing[15] or hydrogen-bond stabilization of the transition state.[16]
The geometry of the diene and dienophile components each propagate into stereochemical details of the product. For intermolecular reactions especially, the preferred positional and stereochemical relationship of substituents of the two components compared to each other are controlled by electronic effects. However, for intramolecular Diels–Alder cycloaddition reactions, the conformational stability of the structure the transition state can be an overwhelming influence.
Regioselectivity
Frontier molecular orbital theory has also been used to explain the regioselectivity patterns observed in Diels–Alder reactions of substituted systems. Calculation of the energy and orbital coefficients of the components' frontier orbitals[17] provides a picture that is in good accord with the more straightforward analysis of the substituents' resonance effects, as illustrated below.

In general, the regioselectivity found for both normal and inverse electron-demand Diels–Alder reaction follows the ortho-para rule, so named, because the cyclohexene product bears substituents in positions that are analogous to the ortho and para positions of disubstituted arenes. For example, in a normal-demand scenario, a diene bearing an electron-donating group (EDG) at C1 has its largest HOMO coefficient at C4, while the dienophile with an electron withdrawing group (EWG) at C1 has the largest LUMO coefficient at C2. Pairing these two coefficients gives the "ortho" product as seen in case 1 in the figure below. A diene substituted at C2 as in case 2 below has the largest HOMO coefficient at C1, giving rise to the "para" product. Similar analyses for the corresponding inverse-demand scenarios gives rise to the analogous products as seen in cases 3 and 4. Examining the canonical mesomeric forms above, it is easy to verify that these results are in accord with expectations based on consideration of electron density and polarization.

In general, with respect to the energetically most well-matched HOMO-LUMO pair, maximizing the interaction energy by forming bonds between centers with the largest frontier orbital coefficients allows the prediction of the main regioisomer that will result from a given diene-dienophile combination.[8] In a more sophisticated treatment, three types of substituents (Z withdrawing: HOMO and LUMO lowering (CF3, NO2, CN, C(O)CH3), X donating: HOMO and LUMO raising (Me, OMe, NMe2), C conjugating: HOMO raising and LUMO lowering (Ph, vinyl)) are considered, resulting in a total of 18 possible combinations. The maximization of orbital interaction correctly predicts the product in all cases for which experimental data is available. For instance, in uncommon combinations involving X groups on both diene and dienophile, a 1,3-substitution pattern may be favored, an outcome not accounted for by a simplistic resonance structure argument.[18] However, cases where the resonance argument and the matching of largest orbital coefficients disagree are rare.
Stereospecificity and stereoselectivity
Diels–Alder reactions, as concerted cycloadditions, are stereospecific. Stereochemical information of the diene and the dienophile are retained in the product, as a syn addition with respect to each component. For example, substituents in a cis (trans, resp.) relationship on the double bond of the dienophile give rise to substituents that are cis (trans, resp.) on those same carbons with respect to the cyclohexene ring. Likewise, cis,cis- and trans,trans-disubstituted dienes give cis substituents at these carbons of the product whereas cis,trans-disubstituted dienes give trans substituents:[19][20]


Diels–Alder reactions in which adjacent stereocenters are generated at the two ends of the newly formed single bonds imply two different possible stereochemical outcomes. This is a stereoselective situation based on the relative orientation of the two separate components when they react with each other. In the context of the Diels–Alder reaction, the transition state in which the most significant substituent (an electron-withdrawing and/or conjugating group) on the dienophile is oriented towards the diene π system and slips under it as the reaction takes place is known as the endo transition state. In the alternative exo transition state, it is oriented away from it. (There is a more general usage of the terms endo and exo in stereochemical nomenclature.)
In cases where the dienophile has a single electron-withdrawing / conjugating substituent, or two electron-withdrawing / conjugating substituents cis to each other, the outcome can often be predicted. In these "normal demand" Diels–Alder scenarios, the endo transition state is typically preferred, despite often being more sterically congested. This preference is known as the Alder endo rule. As originally stated by Alder, the transition state that is preferred is the one with a "maximum accumulation of double bonds." Endo selectivity is typically higher for rigid dienophiles such as maleic anhydride and benzoquinone; for others, such as acrylates and crotonates, selectivity is not very pronounced.[21]
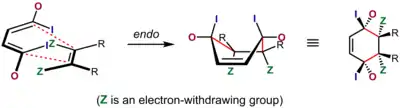
The most widely accepted explanation for the origin of this effect is a favorable interaction between the π systems of the dienophile and the diene, an interaction described as a secondary orbital effect, though dipolar and van der Waals attractions may play a part as well, and solvent can sometimes make a substantial difference in selectivity.[6][22][23] The secondary orbital overlap explanation was first proposed by Woodward and Hoffmann.[24] In this explanation, the orbitals associated with the group in conjugation with the dienophile double-bond overlap with the interior orbitals of the diene, a situation that is possible only for the endo transition state. Although the original explanation only invoked the orbital on the atom α to the dienophile double bond, Salem and Houk have subsequently proposed that orbitals on the α and β carbons both participate when molecular geometry allows.[25]

Often, as with highly substituted dienes, very bulky dienophiles, or reversible reactions (as in the case of furan as diene), steric effects can override the normal endo selectivity in favor of the exo isomer.
The diene
The diene component of the Diels–Alder reaction can be either open-chain or cyclic, and it can host many different types of substituents.[6] It must, however, be able to exist in the s-cis conformation, since this is the only conformer that can participate in the reaction. Though butadienes are typically more stable in the s-trans conformation, for most cases energy difference is small (~2–5 kcal/mol).[26]
A bulky substituent at the C2 or C3 position can increase reaction rate by destabilizing the s-trans conformation and forcing the diene into the reactive s-cis conformation. 2-tert-butyl-buta-1,3-diene, for example, is 27 times more reactive than simple butadiene.[6][27] Conversely, a diene having bulky substituents at both C2 and C3 are less reactive because the steric interactions between the substituents destabilize the s-cis conformation.[27]
Dienes with bulky terminal substituents (C1 and C4) decrease the rate of reaction, presumably by impeding the approach of the diene and dienophile.[28]
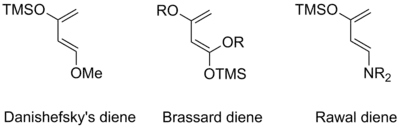
An especially reactive diene is 1-methoxy-3-trimethylsiloxy-buta-1,3-diene, otherwise known as Danishefsky's diene.[29] It has particular synthetic utility as means of furnishing α,β–unsaturated cyclohexenone systems by elimination of the 1-methoxy substituent after deprotection of the enol silyl ether. Other synthetically useful derivatives of Danishefsky's diene include 1,3-alkoxy-1-trimethylsiloxy-1,3-butadienes (Brassard dienes)[30] and 1-dialkylamino-3-trimethylsiloxy-1,3-butadienes (Rawal dienes).[31] The increased reactivity of these and similar dienes is a result of synergistic contributions from donor groups at C1 and C3, raising the HOMO significantly above that of a comparable monosubstituted diene.[3]
Unstable (and thus highly reactive) dienes can be synthetically useful, e.g. o-quinodimethanes, can be generated in situ. In contrast, stable dienes, such as naphthalene, undergo Diels–Alder reactions require forcing conditions and/or highly reactive dienophiles, such as N-phenyl-maleimide. Anthracene, being less aromatic (and therefore more reactive for Diels–Alder syntheses) in its central ring can form a 9,10 adduct with maleic anhydride at 80 °C and even with acetylene, a weak dienophile, at 250 °C.[32]
The dienophile
In a normal demand Diels–Alder reaction, the dienophile has an electron-withdrawing group in conjugation with the alkene; in an inverse-demand scenario, the dienophile is conjugated with an electron-donating group.[9] Dienophiles can be chosen to contain a "masked functionality". The dienophile undergoes Diels–Alder reaction with a diene introducing such a functionality onto the product molecule. A series of reactions then follow to transform the functionality into a desirable group. The end product cannot not be made in a single DA step because equivalent dienophile is either unreactive or inaccessible. An example of such approach is the use of α-chloroacrylonitrile (CH2=CClCN). When reacted with a diene, this dienophile will introduce α-chloronitrile functionality onto the product molecule. This is a "masked functionality" which can be then hydrolyzed to form a ketone. α-Chloroacrylonitrile dienophile is an equivalent of ketene dienophile (CH2=C=O), which would produce same product in one DA step. The problem is that ketene itself cannot be used in Diels–Alder reactions because it reacts with dienes in unwanted manner (by [2+2] cycloaddition), and therefore "masked functionality" approach has to be used.[33] Other such functionalities are phosphonium substituents (yielding exocyclic double bonds after Wittig reaction), various sulfoxide and sulfonyl functionalities (both are acetylene equivalents), and nitro groups (ketene equivalents).[6]
Variants on the classical Diels–Alder reaction
Hetero-Diels–Alder
Diels–Alder reactions involving at least one heteroatom are also known and are collectively called hetero-Diels–Alder reactions.[34] Carbonyl groups, for example, can successfully react with dienes to yield dihydropyran rings, a reaction known as the oxo-Diels–Alder reaction, and imines can be used, either as the dienophile or at various sites in the diene, to form various N-heterocyclic compounds through the aza-Diels–Alder reaction. Nitroso compounds (R-N=O) can react with dienes to form oxazines. Chlorosulfonyl isocyanate can be utilized as a dienophile to prepare Vince lactam.[6][35]
Lewis acid activation
Lewis acids, such as zinc chloride, boron trifluoride, tin tetrachloride, or aluminum chloride, can catalyze the Diels–Alder reactions by binding to the dienophile. Traditionally, the enhanced Diels-Alder reactivity is ascribed to the ability of the Lewis acid to lower the LUMO of the activated dienophile, which results in a smaller normal electron demand HOMO-LUMO orbital energy gap and hence more stabilizing orbital interactions.[36][37][38]
Recent studies, however, have shown that this rationale behind Lewis acid-catalyzed Diels–Alder reactions is incorrect.[39][40][41][42] It is found that Lewis acids accelerate the Diels–Alder reaction by reducing the destabilizing steric Pauli repulsion between the interacting diene and dienophile and not by lowering the energy of the dienophile's LUMO and consequently, enhancing the normal electron demand orbital interaction. The Lewis acid bind via a donor-acceptor interaction to the dienophile and via that mechanism polarizes occupied orbital density away from the reactive C=C double bond of the dienophile towards the Lewis acid. This reduced occupied orbital density on C=C double bond of the dienophile will, in turn, engage in a less repulsive closed-shell-closed-shell orbital interaction with the incoming diene, reducing the destabilizing steric Pauli repulsion and hence lowers the Diels–Alder reaction barrier. In addition, the Lewis acid catalyst also increases the asynchronicity of the Diels–Alder reaction, making the occupied π-orbital located on the C=C double bond of the dienophile asymmetric. As a result, this enhanced asynchronicity leads to an extra reduction of the destabilizing steric Pauli repulsion as well as a diminishing pressure on the reactants to deform, in other words, it reduced the destabilizing activation strain (also known as distortion energy).[43] This working catalytic mechanism is known as Pauli-lowering catalysis,[44] which is operative in a variety of organic reactions.[45][46][47]
The original rationale behind Lewis acid-catalyzed Diels–Alder reactions is incorrect,[39][48][49][50] because besides lowering the energy of the dienophile's LUMO, the Lewis acid also lowers the energy of the HOMO of the dienophile and hence increases the inverse electron demand LUMO-HOMO orbital energy gap. Thus, indeed Lewis acid catalysts strengthen the normal electron demand orbital interaction by lowering the LUMO of the dienophile, but, they simultaneously weaken the inverse electron demand orbital interaction by also lowering the energy of the dienophile's HOMO. These two counteracting phenomena effectively cancel each other, resulting in nearly unchanged orbital interactions when compared to the corresponding uncatalyzed Diels–Alder reactions and making this not the active mechanism behind Lewis acid-catalyzed Diels–Alder reactions.
Asymmetric Diels–Alder
Many methods have been developed for influencing the stereoselectivity of the Diels–Alder reaction, such as the use of chiral auxiliaries, catalysis by chiral Lewis acids,[51] and small organic molecule catalysts.[6] Evans' oxazolidinones,[52] oxazaborolidines,[53][54][55] bis-oxazoline–copper chelates,[56] imidazoline catalysis,[57] and many other methodologies exist for effecting diastereo- and enantioselective Diels–Alder reactions.
Hexadehydro Diels–Alder
In the hexadehydro Diels–Alder reaction, alkynes and diynes are used instead of alkenes and dienes, forming an unstable benzyne intermediate which can then be trapped to form an aromatic product. This reaction allows the formation of heavily functionalized aromatic rings in a single step.[58]
Applications and natural occurrence

The retro-Diels–Alder reaction is used in the industrial production of cyclopentadiene. Cyclopentadiene is a precursor to various norbornenes, which are common monomers. The Diels–Alder reaction is also employed in the production of vitamin B6.
History
The work by Diels and Alder is described in a series of 28 articles published in the Justus Liebigs Annalen der Chemie and Berichte der deutschen chemischen Gesellschaft from 1928 to 1937. The first 19 articles were authored by Diels and Alder, while the later articles were authored by Diels and various contributors.[61][62]
Applications in total synthesis
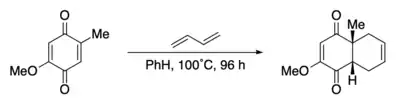
The Diels–Alder reaction was one step in an early preparation of the steroids cortisone and cholesterol.[63] The reaction involved the addition of butadiene to a quinone.
Diels–Alder reactions were used in the original synthesis of prostaglandins F2α and E2.[64] The Diels–Alder reaction establishes the relative stereochemistry of three contiguous stereocenters on the prostaglandin cyclopentane core. Activation by Lewis acidic cupric tetrafluoroborate was required.
.png.webp)
A Diels–Alder reaction was used in the synthesis disodium prephenate,[65] a biosynthetic precursor of the amino acids phenylalanine and tyrosine.
A synthesis of reserpine uses a Diels–Alder reaction to set the cis-decalin framework of the D and E rings.[66]
.png.webp)
In another synthesis of reserpine, the cis-fused D and E rings was formed by a Diels–Alder reaction. Intramolecular Diels–Alder of the pyranone below with subsequent extrusion of carbon dioxide via a retro [4+2] afforded the bicyclic lactam. Epoxidation from the less hindered α-face, followed by epoxide opening at the less hindered C18 afforded the desired stereochemistry at these positions, while the cis-fusion was achieved with hydrogenation, again proceeding primarily from the less hindered face.[67]
.png.webp)
A pyranone was similarly used as the dienophile in the total synthesis of taxol.[68] The intermolecular reaction of the hydroxy-pyrone and α,β–unsaturated ester shown below suffered from poor yield and regioselectivity; however, when directed by phenylboronic acid[69] the desired adduct could be obtained in 61% yield after cleavage of the boronate with 2,2-dimethyl-1,3-propanediol. The stereospecificity of the Diels–Alder reaction in this instance allowed for the definition of four stereocenters that were carried on to the final product.

A Diels–Alder reaction is a key step in the synthesis of (-)-furaquinocin C.[70]
.png.webp)
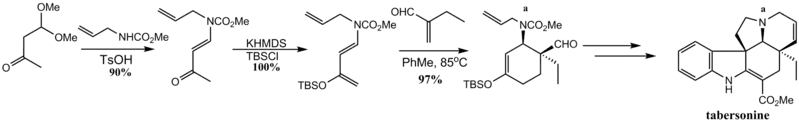
Tabersonine was prepared by a Diels–Alder to establish cis relative stereochemistry of the alkaloid core. Conversion of the cis-aldehyde to its corresponding alkene by Wittig olefination and subsequent ring-closing metathesis with a Schrock catalyst gave the second ring of the alkaloid core. The diene in this instance is notable as an example of a 1-amino-3-siloxybutadiene, otherwise known as a Rawal diene.[71]
(+)-Sterpurene can be prepared by asymmetric D-A reaction[72] that featured a remarkable intramolecular Diels–Alder reaction of an allene. The [2,3]-sigmatropic rearrangement of the thiophenyl group to give the sulfoxide as below proceeded enantiospecifically due to the predefined stereochemistry of the propargylic alcohol. In this way, the single allene isomer formed could direct the Diels-Alder to occur on only one face of the generated 'diene'.

The tetracyclic core of the antibiotic (-)-tetracycline was prepared with a Diels–Alder reaction. Thermally initiated, conrotatory opening of the benzocyclobutene generated the o-quinodimethane, which reacted intermolecularly to give the tetracycline skeleton; the diastereomer shown was then crystallized from methanol after purification by column chromatography. The dienophile's free hydroxyl group is integral to the success of the reaction, as hydroxyl-protected variants did not react under several different reaction conditions.[73]

Takemura et al. synthesized cantharadrin in 1980 by Diels-Alder, utilizing high pressure.[74]
Synthetic applications of the Diels–Alder reaction have been reviewed extensively.[75][76][77][78][79]
See also
- Bradsher cycloaddition
- Wagner-Jauregg reaction
- Imine Diels–Alder reaction
- Aza-Diels–Alder reaction
- Diels-Alderases, enzymes that catalyze Diels–Alder reactions[59]
References
- ↑ Kloetzel, M. C. (1948). "The Diels–Alder Reaction with Maleic Anhydride". Organic Reactions. Vol. 4. pp. 1–59. doi:10.1002/0471264180.or004.01. ISBN 978-0471264187.
- ↑ Holmes, H. L. (1948). "The Diels-Alder Reaction Ethylenic and Acetylenic Dienophiles". Organic Reactions. Vol. 4. pp. 60–173. doi:10.1002/0471264180.or004.02. ISBN 978-0471264187.
- 1 2 Nicolaou, K. C.; Snyder, S. A.; Montagnon, T.; Vassilikogiannakis, G. (2002). "The Diels-Alder Reaction in Total Synthesis". Angewandte Chemie International Edition. 41 (10): 1668–1698. doi:10.1002/1521-3773(20020517)41:10<1668::AID-ANIE1668>3.0.CO;2-Z. PMID 19750686.
- ↑ Atilla Tasdelen, Mehmet (2011). "Diels–Alder "click" reactions: recent applications in polymer and material science". Polymer Chemistry. 2 (10): 2133–2145. doi:10.1039/C1PY00041A.
- ↑ Zweifel, G. S.; Nantz, M. H. (2007). Modern Organic Synthesis: An Introduction. W.H. Freeman and Co. ISBN 978-0-7167-7266-8.
- 1 2 3 4 5 6 7 Carey, Part B., pp. 474–526
- 1 2 Dewar, M. J.; Olivella, S.; Stewart, J. J. (1986). "Mechanism of the Diels-Alder reaction: Reactions of butadiene with ethylene and cyanoethylenes". Journal of the American Chemical Society. 108 (19): 5771–5779. doi:10.1021/ja00279a018. PMID 22175326.
- 1 2 3 Carey, Part A., pp. 836–50
- 1 2 Carey, Part A., p. 839
- ↑ Gajewski, J. J.; Peterson, K. B.; Kagel, J. R. (1987). "Transition-state structure variation in the Diels–Alder reaction from secondary deuterium kinetic isotope effects: The reaction of a nearly symmetrical diene and dienophile is nearly synchronous". Journal of the American Chemical Society. 109 (18): 5545–5546. doi:10.1021/ja00252a052.
- ↑ Houk, K. N.; Lin, Y. T.; Brown, F. K. (1986). "Evidence for the concerted mechanism of the Diels–Alder reaction of butadiene with ethylene". Journal of the American Chemical Society. 108 (3): 554–556. doi:10.1021/ja00263a059. PMID 22175504.
- ↑ Goldstein, E.; Beno, B.; Houk, K. N. (1996). "Density Functional Theory Prediction of the Relative Energies and Isotope Effects for the Concerted and Stepwise Mechanisms of the Diels−Alder Reaction of Butadiene and Ethylene". Journal of the American Chemical Society. 118 (25): 6036–6043. doi:10.1021/ja9601494.
- ↑ Breslow, R.; Guo, T. (1988). "Diels-Alder reactions in nonaqueous polar solvents. Kinetic effects of chaotropic and antichaotropic agents and of β-cyclodextrin". Journal of the American Chemical Society. 110 (17): 5613–5617. doi:10.1021/ja00225a003.
- 1 2 Rideout, D. C.; Breslow, R. (1980). "Hydrophobic acceleration of Diels-Alder reactions". Journal of the American Chemical Society. 102 (26): 7816–7817. doi:10.1021/ja00546a048.
- ↑ Breslow, R.; Rizzo, C. J. (1991). "Chaotropic salt effects in a hydrophobically accelerated Diels–Alder reaction". Journal of the American Chemical Society. 113 (11): 4340–4341. doi:10.1021/ja00011a052.
- ↑ Blokzijl, Wilfried; Engberts, Jan B. F. N. (1992). "Initial-State and Transition-State Effects on Diels–Alder Reactions in Water and Mixed Aqueous Solvents". Journal of the American Chemical Society. 114 (13): 5440–5442. doi:10.1021/ja00039a074.
- ↑ Ashby, E. C.; Chao, L.-C.; Neumann, H. M. (1973). "Organometallic reaction mechanisms. XII. Mechanism of methylmagnesium bromide addition to benzonitrile". Journal of the American Chemical Society. 95 (15): 4896–4904. doi:10.1021/ja00796a022.
- ↑ Fleming, I. (1990). Frontier Orbital and Organic Chemical Reactions. Chichester, UK: Wiley. ISBN 978-0471018193.
- ↑ Kirmse, W.; Mönch, D. (1991). "Umlagerungen von 1,4,4- und 2,2,5-Trimethylbicyclo[3.2.1]oct-6-yl-Kationen". Chemische Berichte. 124 (1): 237–240. doi:10.1002/cber.19911240136.
- ↑ Bérubé, G.; DesLongchamps, P. (1987). "Stéréosélection acyclique-1,5: Synthèse de la chaîne latérale optiquement active de la vitamine E". Bulletin de la Société Chimique de France. 1: 103–115.
- ↑ Houk, K. N.; Luskus, L. J. (1971). "Influence of steric interactions on endo stereoselectivity". Journal of the American Chemical Society. 93 (18): 4606–4607. doi:10.1021/ja00747a052.
- ↑ Kobuke, Y.; Sugimoto, T.; Furukawa, J.; Fueno, T. (1972). "Role of attractive interactions in endo–exo stereoselectivities of Diels–Alder reactions". Journal of the American Chemical Society. 94 (10): 3633–3635. doi:10.1021/ja00765a066.
- ↑ Williamson, K. L.; Hsu, Y.-F. L. (1970). "Stereochemistry of the Diels–Alder reaction. II. Lewis acid catalysis of syn-anti isomerism". Journal of the American Chemical Society. 92 (25): 7385–7389. doi:10.1021/ja00728a022.
- ↑ Woodward, R. B.; Hoffmann, R. (22 October 2013). The conservation of orbital symmetry. Weinheim. ISBN 9781483282046. OCLC 915343522.
{{cite book}}: CS1 maint: location missing publisher (link) - ↑ Wannere, Chaitanya S.; Paul, Ankan; Herges, Rainer; Houk, K. N.; Schaefer, Henry F.; Schleyer, Paul Von Ragué (2007). "The existence of secondary orbital interactions". Journal of Computational Chemistry. 28 (1): 344–361. doi:10.1002/jcc.20532. ISSN 1096-987X. PMID 17109435. S2CID 26096085.
- ↑ Carey, Part A, p. 149
- 1 2 Backer, H. J. (1939). "Le 2,3-Ditertiobutylbutadiène". Recueil des Travaux Chimiques des Pays-Bas. 58 (7): 643–661. doi:10.1002/recl.19390580712.
- ↑ Craig, D.; Shipman, J. J.; Fowler, R. B. (1961). "The Rate of Reaction of Maleic Anhydride with 1,3-Dienes as Related to Diene Conformation". Journal of the American Chemical Society. 83 (13): 2885–2891. doi:10.1021/ja01474a023.
- ↑ Danishefsky, S.; Kitahara, T. (1974). "Useful diene for the Diels–Alder reaction". Journal of the American Chemical Society. 96 (25): 7807–7808. doi:10.1021/ja00832a031.
- ↑ Savard, J.; Brassard, P. (1979). "Regiospecific syntheses of quinones using vinylketene acetals derived from unsaturated esters". Tetrahedron Letters. 20 (51): 4911–4914. doi:10.1016/S0040-4039(01)86747-2.
- ↑ Kozmin, S. A.; Rawal, V. H. (1997). "Preparation and Diels−Alder Reactivity of 1-Amino-3-siloxy-1,3-butadienes". Journal of Organic Chemistry. 62 (16): 5252–5253. doi:10.1021/jo970438q.
- ↑ Margareta Avram (1983). Chimie organica p. 318-323. Editura Academiei Republicii Socialiste România
- ↑ Ranganathan, S.; Ranganathan, D.; Mehrotra, A. K. (1977). "Ketene Equivalents". Synthesis. 1977 (5): 289–296. doi:10.1055/s-1977-24362. S2CID 260335918.
- ↑ Roush, W. R. (1991). "Intramolecular Diels–Alder Reactions". In Trost, B. M.; Flemming, I. (eds.). Comprehensive Organic Synthesis. Vol. 5. pp. 513–550. doi:10.1016/B978-0-08-052349-1.00131-1. ISBN 978-0-08-052349-1.
- ↑ Grieco, P. A.; Larsen, S. D. (1990). "Iminium Ion-Based Diels–Alder Reactions: N-Benzyl-2-Azanorborene" (PDF). Organic Syntheses. 68: 206. doi:10.15227/orgsyn.068.0206.
- ↑ Houk, Kendall N. (1 November 1975). "Frontier molecular orbital theory of cycloaddition reactions". Accounts of Chemical Research. 8 (11): 361–369. doi:10.1021/ar50095a001. ISSN 0001-4842.
- ↑ Fleming, Ian (2009). Molecular orbitals and organic chemical reactions. Chichester, West Sussex, U.K.: Wiley. ISBN 9780470746592.
- ↑ Clayden, Jonathan (2012). Organic chemistry (2nd ed.). Oxford: Oxford University Press. ISBN 9780199270293.
- 1 2 Vermeeren, Pascal; Hamlin, Trevor A.; Fernández, Israel; Bickelhaupt, F. Matthias (6 April 2020). "How Lewis Acids Catalyze Diels–Alder Reactions". Angewandte Chemie International Edition. 59 (15): 6201–6206. doi:10.1002/anie.201914582. PMC 7187354. PMID 31944503.
- ↑ Vermeeren, Pascal; Hamlin, Trevor A.; Fernández, Israel; Bickelhaupt, F. Matthias (2020). "Origin of rate enhancement and asynchronicity in iminium catalyzed Diels–Alder reactions". Chemical Science. 11 (31): 8105–8112. doi:10.1039/D0SC02901G. PMC 8163289. PMID 34094173.
- ↑ Vermeeren, Pascal; Hamlin, Trevor A.; Bickelhaupt, F. Matthias; Fernández, Israel (17 March 2021). "Bifunctional Hydrogen Bond Donor‐Catalyzed Diels–Alder Reactions: Origin of Stereoselectivity and Rate Enhancement". Chemistry: A European Journal. 27 (16): 5180–5190. doi:10.1002/chem.202004496. PMC 8049058. PMID 33169912.
- ↑ Vermeeren, Pascal; Tiezza, Marco Dalla; Dongen, Michelle; Fernández, Israel; Bickelhaupt, F. Matthias; Hamlin, Trevor A. (21 July 2021). "Lewis Acid‐Catalyzed Diels‐Alder Reactions: Reactivity Trends across the Periodic Table". Chemistry: A European Journal. 27 (41): 10610–10620. doi:10.1002/chem.202100522. PMC 8360170. PMID 33780068.
- ↑ Vermeeren, Pascal; Hamlin, Trevor A.; Bickelhaupt, F. Matthias (2021). "Origin of asynchronicity in Diels–Alder reactions". Physical Chemistry Chemical Physics. 23 (36): 20095–20106. Bibcode:2021PCCP...2320095V. doi:10.1039/D1CP02456F. PMC 8457343. PMID 34499069.
- ↑ Hamlin, Trevor A.; Bickelhaupt, F. Matthias; Fernández, Israel (20 April 2021). "The Pauli Repulsion-Lowering Concept in Catalysis" (PDF). Accounts of Chemical Research. 54 (8): 1972–1981. doi:10.1021/acs.accounts.1c00016. hdl:1871.1/a0090b38-9ab8-4c32-9d9a-b3d5de4e5ed3. ISSN 0001-4842. PMID 33759502. S2CID 232337915.
- ↑ Vermeeren, Pascal; Brinkhuis, Francine; Hamlin, Trevor A.; Bickelhaupt, F. Matthias (April 2020). "How Alkali Cations Catalyze Aromatic Diels‐Alder Reactions". Chemistry: An Asian Journal. 15 (7): 1167–1174. doi:10.1002/asia.202000009. PMC 7187256. PMID 32012430.
- ↑ Hansen, Thomas; Vermeeren, Pascal; Yoshisada, Ryoji; Filippov, Dmitri V.; van der Marel, Gijsbert A.; Codée, Jeroen D. C.; Hamlin, Trevor A. (19 February 2021). "How Lewis Acids Catalyze Ring-Openings of Cyclohexene Oxide". The Journal of Organic Chemistry. 86 (4): 3565–3573. doi:10.1021/acs.joc.0c02955. PMC 7901664. PMID 33538169.
- ↑ Tiekink, Eveline H.; Vermeeren, Pascal; Bickelhaupt, F. Matthias; Hamlin, Trevor A. (7 October 2021). "How Lewis Acids Catalyze Ene Reactions". European Journal of Organic Chemistry. 2021 (37): 5275–5283. doi:10.1002/ejoc.202101107. hdl:2066/241097. S2CID 239089361.
- ↑ Vermeeren, Pascal; Hamlin, Trevor A.; Fernández, Israel; Bickelhaupt, F. Matthias (2020). "Origin of rate enhancement and asynchronicity in iminium catalyzed Diels–Alder reactions". Chemical Science. 11 (31): 8105–8112. doi:10.1039/D0SC02901G. PMC 8163289. PMID 34094173.
- ↑ Vermeeren, Pascal; Hamlin, Trevor A.; Bickelhaupt, F. Matthias; Fernández, Israel (17 March 2021). "Bifunctional Hydrogen Bond Donor‐Catalyzed Diels–Alder Reactions: Origin of Stereoselectivity and Rate Enhancement". Chemistry: A European Journal. 27 (16): 5180–5190. doi:10.1002/chem.202004496. PMC 8049058. PMID 33169912.
- ↑ Vermeeren, Pascal; Tiezza, Marco Dalla; Dongen, Michelle; Fernández, Israel; Bickelhaupt, F. Matthias; Hamlin, Trevor A. (21 July 2021). "Lewis Acid‐Catalyzed Diels‐Alder Reactions: Reactivity Trends across the Periodic Table". Chemistry: A European Journal. 27 (41): 10610–10620. doi:10.1002/chem.202100522. PMC 8360170. PMID 33780068.
- ↑ White, James D.; Shaw, Subrata (2011). "cis-2,5-Diaminobicyclo[2.2.2]octane, a New Scaffold for Asymmetric Catalysis via Salen−Metal Complexes". Org. Lett. 13 (9): 2488–91. doi:10.1021/ol2007378. PMID 21462988.
- ↑ Evans, D. A.; Chapman, K. T.; Bisaha, J. (1988). "Asymmetric Diels–Alder cycloaddition reactions with chiral α,β-unsaturated N-acyloxazolidinones". Journal of the American Chemical Society. 110 (4): 1238–1256. doi:10.1021/ja00212a037.
- ↑ Corey, E. J.; Loh, T. P. (1991). "First application of attractive intramolecular interactions to the design of chiral catalysts for highly enantioselective Diels–Alder reactions". Journal of the American Chemical Society. 113 (23): 8966–8967. doi:10.1021/ja00023a066.
- ↑ Corey, E. J.; Shibata, T.; Lee, T. W. (2002). "Asymmetric Diels-Alder reactions catalyzed by a triflic acid activated chiral oxazaborolidine". Journal of the American Chemical Society. 124 (15): 3808–3809. doi:10.1021/ja025848x. PMID 11942799.
- ↑ Ryu, D. H.; Corey, E. J. (2003). "Triflimide activation of a chiral oxazaborolidine leads to a more general catalytic system for enantioselective Diels-Alder addition". Journal of the American Chemical Society. 125 (21): 6388–6390. doi:10.1021/ja035393r. PMID 12785777.
- ↑ Johnson, J. S.; Evans, D. A. (2000). "Chiral bis(oxazoline) copper(II) complexes: Versatile catalysts for enantioselective cycloaddition, Aldol, Michael, and carbonyl Ene reactions". Accounts of Chemical Research. 33 (6): 325–335. doi:10.1021/ar960062n. PMID 10891050.
- ↑ Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. (2000). "New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction". Journal of the American Chemical Society. 122 (17): 4243–4244. doi:10.1021/ja000092s.
- ↑ Hoye, T. R.; Baire, B.; Niu, D.; Willoughby, P. H.; Woods, B. P. Nature, 2012, 490, 208
- 1 2 Minami, Atsushi; Oikawa, Hideaki (2016). "Recent advances of Diels–Alderases involved in natural product biosynthesis". The Journal of Antibiotics. 69 (7): 500–506. doi:10.1038/ja.2016.67. PMID 27301662. S2CID 30482282.
- ↑ Behr, Arno (2000). "Organometallic Compounds and Homogeneous Catalysis". Ullmann's Encyclopedia of Industrial Chemistry. doi:10.1002/14356007.a18_215. ISBN 978-3527306732.
- ↑
- Diels, O.; Alder, K. (1928). "Synthesen in der hydroaromatischen Reihe, I. Mitteilung: Anlagerungen von "Di-en"-kohlenwasserstoffen". Justus Liebigs Annalen der Chemie. 460: 98–122. doi:10.1002/jlac.19284600106.
- Diels, O.; Alder, K. (1929). "Synthesen in der hydroaromatischen Reihe, II. Mitteilung: Über Cantharidin". Berichte der Deutschen Chemischen Gesellschaft. 62 (3): 554–562. doi:10.1002/cber.19290620318.
- Diels, O.; Alder, K. (1929). "Synthesen in der hydroaromatischen Reihe, III. Mitteilung: Synthese von Terpenen, Camphern, hydroaromatischen und heterocyclischen Systemen. Mitbearbeitet von den Herren Wolfgang Lübbert, Erich Naujoks, Franz Querberitz, Karl Röhl, Harro Segeberg". Justus Liebigs Annalen der Chemie. 470: 62–103. doi:10.1002/jlac.19294700106.
- Diels, O.; Alder, K. (1929). "Synthesen in der hydroaromatischen Reihe, IV. Mitteilung: Über die Anlagerung von Maleinsäure-anhydrid an arylierte Diene, Triene und Fulvene (Mitbearbeitet von Paul Pries)". Berichte der Deutschen Chemischen Gesellschaft. 62 (8): 2081–2087. doi:10.1002/cber.19290620829.
- Diels, O.; Alder, K. (1929). "Synthesen in der hydroaromatischen Reihe, V. Über Δ4-Tetrahydro-o-phthalsäure (Stellungnahme zu der Mitteilung von E. H. Farmer und F. L. Warren: Eigenschaften konjugierter Doppelbindungen (VII)". Berichte der Deutschen Chemischen Gesellschaft. 62 (8): 2087–2090. doi:10.1002/cber.19290620830.
- Diels, O.; Alder, K. (1929). "Synthesen in der hydroaromatischen Reihe, VI. Mitteilung, Kurt Alder und Gerhard Stein: Über partiell hydrierte Naphtho- und Anthrachinone mit Wasserstoff in γ- bzw. δ-Stellung. (Mitbearbeitet von Paul Pries und Hans Winckler)". Berichte der Deutschen Chemischen Gesellschaft. 62 (8): 2337–2372. doi:10.1002/cber.19290620872.
- Diels, O.; Alder, K. (1930). "Synthesen in der hydroaromatischen Reihe, VII. Mitteilung. (Mitbearbeitet von den Harren Ernst Petersen und Franz Querberitz.)". Justus Liebigs Annalen der Chemie. 478: 137–154. doi:10.1002/jlac.19304780109.
- Diels, O.; Alder, K. (1931). "Synthesen in der hydroaromatischen Reihe, VIII. Mitteilung: Dien-Synthesen des Anthracens. Anthracen-Forme". Justus Liebigs Annalen der Chemie. 486: 191–202. doi:10.1002/jlac.19314860110.
- Diels, O.; Alder, K. (1931). "Synthesen in der hydroaromatischen Reihe, IX. Mitteilung: Synthese des Camphenilons und des Santens". Justus Liebigs Annalen der Chemie. 486: 202–210. doi:10.1002/jlac.19314860111.
- Diels, O.; Alder, K. (1931). "Synthesen in der hydroaromatischen Reihe, X. Mitteilung: "Dien-Synthesen"︁ mit Pyrrol und seinen Homologen". Justus Liebigs Annalen der Chemie. 486: 211–225. doi:10.1002/jlac.19314860112.
- Diels, O.; Alder, K. (1931). "Synthesen in der hydroaromatischen Reihe, XI. Mitteilung. ("Dien-Synthesen"︁ des Cyclopentadiens, Cyclo-hexadiens und Butadiens mit Acetylen-dicarbonsäure und ihren Estern". Justus Liebigs Annalen der Chemie. 490: 236–242. doi:10.1002/jlac.19314900109.
- Diels, O.; Alder, K. (1931). "Synthesen in der hydroaromatischen Reihe, XII. Mitteilung. ("Dien-Synthesen"︁ sauerstoffhaltiger Heteroringe. 2. Dien-Synthesen des Furans.)". Justus Liebigs Annalen der Chemie. 490: 243–257. doi:10.1002/jlac.19314900110.
- Diels, O.; Alder, K. (1931). "Synthesen in der hydroaromatischen Reihe, XIII. Mitteilung. ("Dien-Synthesen"︁ sauerstoffhaltiger Heteroringe. 3. Dien-Synthesen der Cumaline.)". Justus Liebigs Annalen der Chemie. 490: 257–266. doi:10.1002/jlac.19314900111.
- Diels, O.; Alder, K. (1931). "Synthesen in der hydroaromatischen Reihe, XIV. Mitteilung. ("Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 2. Dien-Synthesen der Pyrrole mit Acetylen-dicarbonsäure und mit ihren Estern.)". Justus Liebigs Annalen der Chemie. 490: 267–276. doi:10.1002/jlac.19314900112.
- Diels, O.; Alder, K. (1931). "Synthesen in der hydroaromatischen Reihe, XV. Mitteilung. ("Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 3. Dien-Synthesen der Indole.)". Justus Liebigs Annalen der Chemie. 490: 277–294. doi:10.1002/jlac.19314900113.
- Diels, O.; Alder, K. (1932). "Synthesen in der hydroaromatischen Reihe, XVI. Mitteilung. ("Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 4. Dien-Synthesen der Pyrrole, Imidazole und Pyrazole.)". Justus Liebigs Annalen der Chemie. 498: 1–15. doi:10.1002/jlac.19324980102.
- Diels, O.; Alder, K. (1932). "Synthesen in der hydroaromatischen Reihe, XVII. Mitteilung. ("Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 5. Dien-Synthesen des Pyridins, Chinolins, Chinaldins und Isochinolins.)". Justus Liebigs Annalen der Chemie. 498: 16–49. doi:10.1002/jlac.19324980103.
- Diels, O.; Alder, K. (1933). "Synthesen in der hydroaromatischen Reihe, XVIII "Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 6. Dien-Synthesen des Pyridins. Zur Kenntnis des Chinolizins, Indolizins, Norlupinans und Pseudolupinins". Justus Liebigs Annalen der Chemie. 505: 103–150. doi:10.1002/jlac.19335050109.
- Diels, O.; Alder, K. (1934). "Synthesen in der hydroaromatischen Reihe, XIX. "Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 7. Zur Kenntnis der Primärprodukte bei den Dien-Synthesen des Pyridins, Chinolins und Chinaldins". Justus Liebigs Annalen der Chemie. 510: 87–128. doi:10.1002/jlac.19345100106.
- Diels, O.; Reese, J. (1934). "Synthesen in der hydroaromatischen Reihe, XX. Über die Anlagerung von Acetylen-dicarbonsäureester an Hydrazobenzol". Justus Liebigs Annalen der Chemie. 511: 168–182. doi:10.1002/jlac.19345110114.
- Diels, O.; Meyer, R. (1934). "Synthesen in der hydroaromatischen Reihe, XXI. "Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 8. Über den Verlauf der Dien-Synthese des Pyridins in methylalkoholischer Lösung". Justus Liebigs Annalen der Chemie. 513: 129–145. doi:10.1002/jlac.19345130108.
- Diels, O.; Friedrichsen, W. (1934). "Synthesen in der hydroaromatischen Reihe, XXII. Über die Anthracen–C4O3-Addukte, ihre Eignung zu Dien-Synthesen und ein neues Prinzip zur Synthese von Phtalsäuren und Dihydro-phtalsäuren". Justus Liebigs Annalen der Chemie. 513: 145–155. doi:10.1002/jlac.19345130109.
- Diels, O.; Möller, F. (1935). "Synthesen in der hydroaromatischen Reihe, XXIII. "Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 9. Stilbazol und Acetylen-dicarbonester". Justus Liebigs Annalen der Chemie. 516: 45–61. doi:10.1002/jlac.19355160104.
- Diels, O.; Kech, H. (1935). "Synthesen in der hydroaromatischen Reihe, XXIV "Dien-Synthesen"︁ stickstoffhaltiger Heteroringe". Justus Liebigs Annalen der Chemie. 519: 140–146. doi:10.1002/jlac.19355190112.
- Diels, O.; Reese, J. (1935). "Synthesen in der hydroaromatischen Reihe, XXV Über die Addukte aus Acetylen-dicarbonsäureester und Hydrazo-Verbindungen (2)". Justus Liebigs Annalen der Chemie. 519: 147–157. doi:10.1002/jlac.19355190113.
- Diels, O.; Harms, J. (1935). "Synthesen in der hydroaromatischen Reihe, XXVI. "Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 11. Über die aus Isochinolin und Acetylen-dicarbonsäureester entstehenden Addukte". Justus Liebigs Annalen der Chemie. 525: 73–94. doi:10.1002/jlac.19365250107.
- Diels, O.; Schrum, H. (1937). "Synthesen in der hydroaromatischen Reihe,XXVII. "Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 12. Über den Abbau der "gelben Substanz"︁ zu einem Isomeren des Norlupinans (1-Methyl-octahydro-indolizin)". Justus Liebigs Annalen der Chemie. 530: 68–86. doi:10.1002/jlac.19375300106.
- Diels, O.; Pistor, H. (1937). "Synthesen in der hydroaromatischen Reihe, XXVIII. "Dien-Synthesen"︁ stickstoffhaltiger Heteroringe. 13. α-Picolin und Acetylen-dicarbonsäureeste". Justus Liebigs Annalen der Chemie. 530: 87–98. doi:10.1002/jlac.19375300107.
- ↑ "The Nobel Prize in Chemistry 1950". The Nobel Foundation. Retrieved 19 February 2016.
- ↑ Woodward, R. B.; Sondheimer, F.; Taub, D.; Heusler, K.; McLamore, W. M. (1952). "The Total Synthesis of Steroids". Journal of the American Chemical Society. 74 (17): 4223–4251. doi:10.1021/ja01137a001.
- ↑ Corey, E. J.; Weinshenker, N. M.; Schaaf, T. K.; Huber, W. (1969). "Stereo-controlled synthesis of prostaglandins F-2a and E-2 (dl)". Journal of the American Chemical Society. 91 (20): 5675–7. doi:10.1021/ja01048a062. PMID 5808505.
- ↑ Danishefsky, S.; Hirama, M.; Fritsch, N.; Clardy, J. (1979). "Synthesis of disodium prephenate and disodium epiprephenate. Stereochemistry of prephenic acid and an observation on the base-catalyzed rearrangement of prephenic acid to p-hydroxyphenyllactic acid". Journal of the American Chemical Society. 101 (23): 7013–7018. doi:10.1021/ja00517a039.
- ↑ Wender, P. A.; Schaus, J. M.; White, A. W. (1980). "General methodology for cis-hydroisoquinoline synthesis: Synthesis of reserpine". Journal of the American Chemical Society. 102 (19): 6157–6159. doi:10.1021/ja00539a038.
- ↑ Martin, S. F.; Rueger, H.; Williamson, S. A.; Grzejszczak, S. (1987). "General strategies for the synthesis of indole alkaloids. Total synthesis of (±)-reserpine and (±)-α-yohimbine". Journal of the American Chemical Society. 109 (20): 6124–6134. doi:10.1021/ja00254a036.
- ↑ Nicolaou, K. C.; Yang, Z.; Liu, J. J.; Ueno, H.; Nantermet, P. G.; Guy, R. K.; Claiborne, C. F.; Renaud, J.; Couladouros, E. A.; Paulvannan, K.; Sorensen, E. J. (1994). "Total synthesis of taxol". Nature. 367 (6464): 630–4. Bibcode:1994Natur.367..630N. doi:10.1038/367630a0. PMID 7906395. S2CID 4371975.
- ↑ Narasaka, K.; Shimada, S.; Osoda, K.; Iwasawa, N. (1991). "Phenylboronic Acid as a Template in the Diels-Alder Reaction". Synthesis. 1991 (12): 1171–1172. doi:10.1055/s-1991-28413.
- ↑ Smith, A. B.; Sestelo, J. P.; Dormer, P. G. (1995). "Total Synthesis of (−)-Furaquinocin C". Journal of the American Chemical Society. 117 (43): 10755–10756. doi:10.1021/ja00148a023.
- ↑ Kozmin, S. A.; Rawal, V. H. (1998). "A General Strategy to Aspidosperma Alkaloids: Efficient, Stereocontrolled Synthesis of Tabersonine". Journal of the American Chemical Society. 120 (51): 13523–13524. doi:10.1021/ja983198k.
- ↑ Gibbs, R. A.; Okamura, W. H. (1988). "A short enantioselective synthesis of (+)-sterpurene: Complete intramolecular transfer of central to axial to central chiral elements". Journal of the American Chemical Society. 110 (12): 4062–4063. doi:10.1021/ja00220a069.
- ↑ Charest, M. G.; Siegel, D. R.; Myers, A. G. (2005). "Synthesis of (-)-tetracycline". Journal of the American Chemical Society. 127 (23): 8292–3. doi:10.1021/ja052151d. PMID 15941256.
- ↑ Dauben, W. G.; Kessel, C. R.; Takemura, K. H. (1980). "Simple, efficient total synthesis of cantharidin via a high-pressure Diels–Alder reaction". Journal of the American Chemical Society. 102 (22): 6893–6894. doi:10.1021/ja00542a060.
- ↑ Holmes, H. L. (1948). "The Diels–Alder Reaction Ethylenic and Acetylenic Dienophiles". Organic Reactions. Vol. 4. pp. 60–173. doi:10.1002/0471264180.or004.02. ISBN 978-0471264187.
- ↑ Butz, L. W.; Rytina, A. W. (1949). "The Diels–Alder Reaction Quinones and Other Cyclenones". Organic Reactions. Vol. 5. pp. 136–192. doi:10.1002/0471264180.or005.03. ISBN 978-0471264187.
- ↑ Kloetzel, M. C. (1948). "The Diels–Alder Reaction with Maleic Anhydride". Organic Reactions. Vol. 4. pp. 1–59. doi:10.1002/0471264180.or004.01. ISBN 978-0471264187.
- ↑ Heintzelman, G. R.; Meigh, I. R.; Mahajan, Y. R.; Weinreb, S. W. (2005). "Diels-Alder Reactions of Imino Dienophiles". Organic Reactions. Vol. 65. pp. 141–599. doi:10.1002/0471264180.or065.02. ISBN 978-0471264187.
- ↑ Ciganek, E. (1984). "The Intramolecular Diels-Alder Reaction". Organic Reactions. Vol. 32. pp. 1–374. doi:10.1002/0471264180.or032.01. ISBN 978-0471264187.
Bibliography
- Carey, Francis A.; Sundberg, Richard J. (2007). Advanced Organic Chemistry: Part B: Reactions and Synthesis (5th ed.). New York: Springer. ISBN 978-0387683546.