Carbonate-associated sulfates (CAS) are sulfate species found in association with carbonate minerals, either as inclusions, adsorbed phases, or in distorted sites within the carbonate mineral lattice.[1][2][3][4][5][6][7][8] It is derived primarily from dissolved sulfate in the solution from which the carbonate precipitates. In the ocean, the source of this sulfate is a combination of riverine and atmospheric inputs, as well as the products of marine hydrothermal reactions and biomass remineralisation.[9][10][11][12][13] CAS is a common component of most carbonate rocks, having concentrations in the parts per thousand within biogenic carbonates and parts per million within abiogenic carbonates.[14][15][16][17] Through its abundance and sulfur isotope composition, it provides a valuable record of the global sulfur cycle across time and space.
Importance of sulfur (and CAS) to biogeochemistry
Sulfur compounds play a major role in global climate, nutrient cycling, and the production and distribution of biomass. They can have significant effects on cloud formation and greenhouse forcing, and their distribution responds to the oxidation state of the atmosphere and oceans, as well as the evolution of different metabolic strategies. We can resolve the response of sulfur to biogeochemical change by measuring the abundance and isotopic composition of different sulfur species in different environments at different times.
But how do abundance and isotopic composition of different sulfur reservoirs inform our understanding of biogeochemical processes? The oxidation and reduction of sulfur species often involves the breakage or formation of chemical bonds involving S atoms. Because the thermodynamic stability of certain bonds is often greater when they involve heavier isotopes, an oxidation or reduction reaction can enrich the reactant pool (reservoir) or product pool in compounds containing the heavier isotope, relative to each other. This is known as an isotope effect. The extent to which such a mass-dependent reaction operates in the world's oceans or atmosphere determines how much heavier or lighter various reservoirs of sulfur species will become.
The largest sulfur pool on Earth is that of marine or "seawater" sulfate. Traditionally, the isotopic composition of seawater sulfate is obtained by analysis of sulfate minerals within evaporites, which are somewhat sparse in the geologic record, often poorly preserved, and necessarily associated with complicated and excursive events such as local sea level change.[18][19][20][21][22][23][24][25] Marine barites are similarly limited.[26][27][28][29][30] Carbonate-associated sulfate (CAS) provides geochemists with a more ubiquitous source of material for the direct measurement of seawater sulfate, provided the degree of secondary alteration and diagenetic history of the carbonate and CAS can be constrained.
Sulfate and the global sulfur cycle
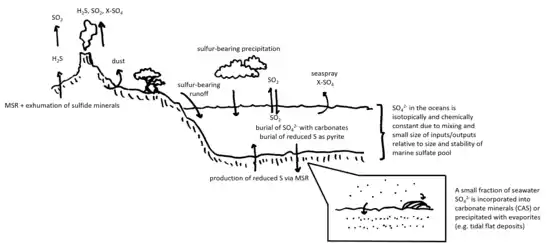
Earth's sulfur cycle is complex. Volcanoes release both reduced and oxidized sulfur species into the atmosphere, where they are further oxidized by reaction with oxygen to SO2 and various sulfates. These oxidized sulfur species enter groundwater and the oceans both directly (rain/snow) or by incorporation into biomass, which decays to sulfates and sulfides, again by a combination of biological and abiological processes.[31][32] Some of this sulfate is reduced through microbial metabolism (microbial sulfate reduction or MSR) or by hydrothermal processes, yielding sulfides, thiosulfate, and elemental sulfur. Some reduced sulfur species are buried as metal-sulfide compounds, some are cyclically reduced and oxidized in the oceans and sediments indefinitely, and some are oxidized back into sulfate minerals, which precipitate out in tidal flats, lakes, and lagoons as evaporite deposits or are incorporated into the structure of carbonate and phosphate minerals in the ocean (i.e. as CAS).[33][34][35][36][37][38][39]
Because oxidation-reduction reactions with sulfur species are often accompanied by mass-dependent fractionation, the sulfur isotope composition of the various pools of reduced and oxidized sulfur species in the water column, sediment, and rock record is a clue to how sulfur moves between those pools or has moved in the past. For example, sulfur at the time of Earth's formation should have (barring some accretion-related fractionation process for which there is little evidence) had a δ34S value of about 0‰, while sulfate in the modern oceans (the dominant marine sulfur species) has a δ34S of about +21‰.[40][41][6] This implies that, over geologic time, a reservoir of correspondingly depleted (i.e. 34S-poor) sulfur was buried in the crust and possibly subducted into the deep mantle. This is because sulfate's reduction to sulfide is typically accompanied by a negative isotope effect, which (depending on the sulfate-reducing microorganism's enzymatic machinery, temperature, and other factors) can be tens of per mille.[42][43][44][45][46] This effect can be compounded through sulfur disproportionation, a process by which some microbes reduce sulfate to sulfides and thiosulfate, both of which can be 34S-depleted by tens of per mille relative to the starting sulfate pool.[47][48] Depleted sulfides and thiosulfate can then be repeatedly oxidized and reduced again, until the final, total sulfide pool that is measured has δ34S values of -70 or -80‰.[49][47][48][50] The formation of a "lighter" S-isotope pool leaves behind an enriched pool, and so the enrichment of seawater sulfate is taken as evidence that some large amount of reduced sulfur (in the form, perhaps, of metal-sulfide minerals) was buried and incorporated into the crust.
Recording seawater sulfate

Carbonate-associated sulfate (CAS) represents a small fraction of seawater sulfate, buried (and to some extent, preserved) with carbonate sediments. Thus, the changing δ34S value of CAS through time should theoretically scale with the changing amount of reduced sulfur species being buried as metal-sulfides and the correspondingly enriched ocean. The enrichment of marine sulfate in 34S should in turn scale with things like: the level of oxygen in the oceans and atmosphere, the initial appearance and proliferation of sulfur-reducing metabolisms among the world's microbial communities, and perhaps local-scale climate events and tectonism.[51][52][37] The more positive the δ34S of marine sulfate, the more sulfate reduction and/or burial/removal of reduced, 34S-depleted sulfur species must be occurring.
There are some limitations, however, to the use of carbonate-associated sulfate's isotopic composition as a proxy for the isotopic composition of marine sulfate (and thus as a proxy for the response of the sulfur cycle to major climatological and geobiological events) through time. First, there is the question of: how representative is a particular carbonate rock's CAS of marine sulfate at the time of the rock's deposition? Various diagenetic processes (meaning: deformation by burial and exhumation, exposure to groundwater and meteoric fluids carrying sulfur species from more modern sources, etc.) can alter the abundance and isotopic composition of CAS.[53] And so, carbonate mineral crystals used as a sulfur cycle proxy must be carefully selected to avoid highly altered or recrystallized material.
Significant to this problem is the position that carbonate-associated sulfate occupies in the structure of carbonate minerals. X-ray diffraction and reflectance spectroscopy have revealed how the replacement of the carbonate group with sulfate ion tetrahedra expands the crystal lattice. (It follows that higher Mg-content in the carbonate, which itself depends on the ocean's weathering inputs, pH, etc. and increases the distortion of the crystal lattice and rock volume, can also allow for the incorporation of more sulfate into the mineral structure.) Any processes that further distort the crystal lattice can cause sulfate to be lost from or added to the carbonate mineral, possibly overprinting the marine sulfate signal from the time of deposition.[54][55][56][57][58]
On balance, CAS preserves and records the isotopic composition of seawater sulfate at the time of its deposition, provided the host carbonate has not been completely recrystallized or undergone replacement via sulfur-bearing fluids after burial. If the host carbonate has been altered in this way, CAS may contain a mixture of signals that is difficult to characterize.
Measuring
Measuring abundance
In measuring the abundance and isotopic composition of CAS, it is important to know exactly what is being measured: CAS within particular shell fragments, corals, microbialites, cements, or otherwise. The first step is therefore to separate out the desired component for measurement. This could mean drilling and powdering a rock (if the CAS measurement of the whole rock is desired) or sorting sediments by visual identification of particular microfossils or mineral phases, using fine tweezers and drills under a microscope. The fragments, sediments, or powders should be cleaned (likely by sonication) and exposed only to deionized and filtered water, so that no contaminant sulfur species are introduced, and the original CAS is not further reduced, oxidized, or otherwise altered. Next, the clean samples must be measured.
In one method, these samples are "digested" in an acid, likely HCl, which will liberate CAS from inclusions or the mineral lattice by dissolving the calcite mineral. The resulting sulfate ions are precipitated (often by mixture with barium chloride to produce barium sulfate), and the solid sulfate precipitate is filtered, dried, and transferred to an elemental analysis pipeline, which may involve the combustion of the sample and the mass balance of its various combustion products (which should include CO2 and SO2). Knowledge of the ratio of sulfur to oxygen and other components in the elemental analysis pipeline allows one to calculate the amount of sulfate introduced to the pipeline by the sample. This, along with the precise measurement of the original sample's mass and volume, yields a sulfate concentration for the original sample.[6][59] The "combustion" and reaction to SO2 can also bypassed by instead passing the acid-dissolved sample through an ion chromatography column, wherein different ions' polarity determines the strength of their interactions with polymers in the column, such that they are retained in the column for different amounts of time.
The concentration of CAS may also be measured by spectroscopic methods. This could mean using the characteristic X-ray-induced fluorescence of sulfur, oxygen, carbon, and other elements in the sample to determine the abundance and ratios of each component, or the energy spectrum of an electron beam transmitted through the sample.
It is also important to calibrate your measurement using standards of a known sulfate concentration, so that the strength/intensity of the signal associated with each sample can be mapped to a particular abundance.
Measuring isotopic composition
The abundance of CAS in a particular sample depends as much on the circumstances of a particular carbonate rock's formation and diagenetic history as it does on the processes acting on the marine sulfate pool that generated it. Thus, it is important to have both the abundance/concentration of CAS in a sample and its isotopic composition to understand its place in the marine sulfate record. As mentioned above, different biogeochemical processes produce different isotope effects under equilibrium and disequilibrium conditions: microbial sulfur reduction and sulfur disproportionation can produce equilibrium and kinetic isotope effects of many 10s of per mille. The sulfur isotope composition of the ocean (or a lake, lagoon, or other body) is critical to understanding the extent to which those processes controlled the global sulfur cycle throughout the past. Just as the carbon and oxygen isotope composition of the carbonate host rock can illuminate temperature and local climate history, the sulfur and oxygen isotope composition of CAS can illuminate the cause and effect relationships between that history and the sulfur cycle. Isotopic composition of CAS and carbonate host rock can both be measured by "elemental analysis" wherein sulfate or carbonate is "burned" or otherwise volatilized and the ionized isotopes are accelerated along a path, the length and duration of which is a function of their masses. The ratio of different isotopes to one another is assessed by comparison to blanks and standards. However, SO2, the analyte used in this method, presents some difficulties as the isotopic composition of the component oxygen may also vary, affecting the mass measurement. SO2 can also "stick" to or react with other compounds in the mass spectrometer line. Thus, if high precision is needed, sulfate samples are reduced to sulfides, which are then fluorinated to produce the inert and stable-isotopologue-free compound SF6, which can be passed through a specialized mass spectrometer. These methods, mass spectrometry and clumped isotope mass spectrometry, are discussed in greater detail in their primary articles.[60][6][59][8]
The isotopic composition of CAS is often discussed in terms of δ34S, which is a way of expressing the ratio of the isotope 34S to 32S in a sample, relative to a standard such as the Canyon Diablo Troilite. δ34S (expressed in ‰) is equal to . The isotope effect of a particular process (microbial sulfate reduction, for example) is often expressed as an ε value (also in ‰) which refers to the difference in the δ value of the reactant pool and the product pool.
![{\textstyle \left[{\frac {(^{34}S/^{32}S)_{sample}}{(^{34}S/^{32}S)_{standard}}}-1\right]*1000}](../I/852014247aafcb99c44743aee9884a741b2e03ac.svg)
While studies of the sulfur isotope composition of seawater sulfate, CAS, marine barite, and evaporites typically discuss the relative 34S enrichment of depletion of these pools, there are other minor but stable isotopes of sulfur that can also be measured, though to lower precision given their rarity. These include 33S and 36S. Mass-dependent and mass-independent fractionation of minor sulfur isotopes may also be an important gauge for the sulfur cycle through geologic time. 33S and 36S must, however, be measured at high-precision via fluorination to SF6 before passing through a mass spectrometer.
Interpreting measurements
Interpreting the sulfur isotope composition of CAS can be complex. As discussed above, if seawater sulfate at a particular horizon in the geologic record gets heavier (i.e. more enriched in 34S relative to seawater sulfate before it) that could mean that the 34S-depleted products of sulfur-reducing reactions are being buried as sulfide minerals and removed from the oceans, possibly because of an instance of ocean anoxia[61][62][63] or an increase in dissimilatory sulfate reduction by marine microorganisms. But it could also mean that the CAS measured at that particular horizon was derived not from seawater sulfate at the time of carbonate deposition, but from fluids moving through the sediment or porous rock from a later time, in which sulfate could have been enriched by processes in a more oxidizing world. It could mean that there is a hitherto uncharacterized kinetic isotope effect associated with the incorporation of sulfate into a particular carbonate texture (shrubs vs. nodules vs. acicular cements vs. other conformations). Distinguishing between the effects of true changes in ancient ocean dynamics/chemistry and the effects of early- and late-stage diagenesis on CAS isotope composition is possible only through careful analyses that: compare the CAS record to the seawater sulfate record preserved in evaporites and marine barite, and carefully screen samples for their thermodynamic stability and evidence of alteration.[3][64][4] Such samples could include brachiopod shell fragments (which are made of stable, low-Mg calcite that visibly resists alteration after cementation).[4][65][66][8][67]
Some important insights from CAS studies
The CAS record can preserve evidence of major changes in oxidation state of the ocean in response to climate. For example, the Great Oxygenation Event led to the oxidation of reduced sulfur species, increasing the flux of sulfate into the oceans. This led to a corresponding depletion of 34S in the marine sulfate pool — a depletion recorded in the sulfur isotope composition of marginal marine evaporite deposits and CAS in marine carbonates.[51][52][37]
Before the Great Oxygenation Event, when atmospheric and marine oxygen was low, it is expected that oxidized sulfur species like sulfate would have been much less abundant. Exactly how much less may be estimated from the δ34S value of sediments in modern analog environments like anoxic lakes, and their comparison to preserved Archean-age seawater sulfate (as found in CAS).[68]
The Great Oxygenation Event lead not just to the oxygenation of Earth's oceans, but to the development of the ozone layer. Prior to this, the Archean Earth was exposed to high-energy radiation that caused mass-independent fractionation of various pools, including sulfur (which would lead to an expected negative δ34S excursion in the marine sulfate pool). The marine sulfate record preserved in CAS complicates this view, as late or Neo-Archean CAS samples seem to have positive δ34S.[59]
The CAS record may (or may not) preserve evidence of the rise of microbial sulfate reduction, in the form of a negative δ34S excursion between 2.7 and 2.5 Ga.[69][70]
The variation in sulfur isotope composition of sulfate associated with the different components of a carbonate or phosphate rock may also provide insights into the diagenetic history of a sample and the degree of preservation of the original texture and chemistry in different types of grains.[37][8]
Ongoing improvements to CAS studies
Much of the ongoing work in the field of carbonate-associated sulfate is dedicated to characterizing sources of variation in the CAS record, answering questions like: how are sulfate ions incorporated into the mineral structure of different Ca-carbonate and Ca-Mg-carbonate morphotypes, mechanistically speaking? And which morphotypes are most likely to contain CAS derived from primary marine sulfate?
Just as for other geochemical proxies, the utility and reliability of CAS measurements will improve with the advent of more sensitive measurement techniques, and the characterization of more isotope standards.
References
- ↑ Kaplan, I.R.; Emery, K.O.; Rittenbebg, S.C. (April 1963). "The distribution and isotopic abundance of sulphur in recent marine sediments off southern California". Geochimica et Cosmochimica Acta. 27 (4): 297–331. Bibcode:1963GeCoA..27..297K. doi:10.1016/0016-7037(63)90074-7.
- ↑ Makhitiyeva, V (1974). "Sulfur isotopic composition of fossil molluscan shells as an indicator of hydrochemical conditions in ancient basins". Geochemistry International. 11: 1188–1192.
- 1 2 W. Burdett, James; A. Arthur, Michael; Richardson, Mark (September 1989). "A Neogene seawater sulfur isotope age curve from calcareous pelagic microfossils". Earth and Planetary Science Letters. 94 (3–4): 189–198. Bibcode:1989E&PSL..94..189B. doi:10.1016/0012-821X(89)90138-6.
- 1 2 3 Kampschulte, A; Strauss, H (April 2004). "The sulfur isotopic evolution of Phanerozoic seawater based on the analysis of structurally substituted sulfate in carbonates". Chemical Geology. 204 (3–4): 255–286. Bibcode:2004ChGeo.204..255K. doi:10.1016/j.chemgeo.2003.11.013.
- ↑ Amend, Jan P.; Edwards, Katrina J.; Lyons, Timothy W. (2004). Sulfur Biogeochemistry: Past and Present. Geological Society of America. ISBN 9780813723792.
- 1 2 3 4 Paris, Guillaume; Sessions, Alex L.; Subhas, Adam V.; Adkins, Jess F. (May 2013). "MC-ICP-MS measurement of δ34S and ∆33S in small amounts of dissolved sulfate". Chemical Geology. 345: 50–61. Bibcode:2013ChGeo.345...50P. doi:10.1016/j.chemgeo.2013.02.022.
- ↑ Paris, Guillaume; Fehrenbacher, Jennifer S.; Sessions, Alex L.; Spero, Howard J.; Adkins, Jess F. (April 2014). "Experimental determination of carbonate-associated sulfate δ S in planktonic foraminifera shells" (PDF). Geochemistry, Geophysics, Geosystems. 15 (4): 1452–1461. Bibcode:2014GGG....15.1452P. doi:10.1002/2014GC005295.
- 1 2 3 4 Present, Theodore M.; Paris, Guillaume; Burke, Andrea; Fischer, Woodward W.; Adkins, Jess F. (December 2015). "Large Carbonate Associated Sulfate isotopic variability between brachiopods, micrite, and other sedimentary components in Late Ordovician strata" (PDF). Earth and Planetary Science Letters. 432: 187–198. Bibcode:2015E&PSL.432..187P. doi:10.1016/j.epsl.2015.10.005. hdl:10023/9759.
- ↑ Cuif, Jean-Pierre; Dauphin, Yannicke; Doucet, Jean; Salome, Murielle; Susini, Jean (January 2003). "XANES mapping of organic sulfate in three scleractinian coral skeletons". Geochimica et Cosmochimica Acta. 67 (1): 75–83. Bibcode:2003GeCoA..67...75C. doi:10.1016/S0016-7037(02)01041-4.
- ↑ Dauphin, Y. (1 November 2005). "Speciation and distribution of sulfur in a mollusk shell as revealed by in situ maps using X-ray absorption near-edge structure (XANES) spectroscopy at the S K-edge". American Mineralogist. 90 (11–12): 1748–1758. Bibcode:2005AmMin..90.1748D. doi:10.2138/am.2005.1640. S2CID 95539399.
- ↑ Cusack, Maggie; Dauphin, Yannicke; Cuif, Jean-Pierre; Salomé, Murielle; Freer, Andy; Yin, Huabing (August 2008). "Micro-XANES mapping of sulphur and its association with magnesium and phosphorus in the shell of the brachiopod, Terebratulina retusa". Chemical Geology. 253 (3–4): 172–179. Bibcode:2008ChGeo.253..172C. doi:10.1016/j.chemgeo.2008.05.007.
- ↑ Balan, Etienne; Aufort, Julie; Pouillé, Sophie; Dabos, Marie; Blanchard, Marc; Lazzeri, Michele; Rollion-Bard, Claire; Blamart, Dominique (26 June 2017). "Infrared spectroscopic study of sulfate-bearing calcite from deep-sea bamboo coral" (PDF). European Journal of Mineralogy. 29 (3): 397–408. Bibcode:2017EJMin..29..397B. doi:10.1127/ejm/2017/0029-2611.
- ↑ Perrin, J.; Rivard, C.; Vielzeuf, D.; Laporte, D.; Fonquernie, C.; Ricolleau, A.; Cotte, M.; Floquet, N. (January 2017). "The coordination of sulfur in synthetic and biogenic Mg calcites: The red coral case". Geochimica et Cosmochimica Acta. 197: 226–244. Bibcode:2017GeCoA.197..226P. doi:10.1016/j.gca.2016.10.017.
- ↑ W. Burdett, James; A. Arthur, Michael; Richardson, Mark (1989-09-01). "A Neogene seawater sulfur isotope age curve from calcareous pelagic microfossils". Earth and Planetary Science Letters. 94 (3): 189–198. Bibcode:1989E&PSL..94..189B. doi:10.1016/0012-821X(89)90138-6. ISSN 0012-821X.
- ↑ Kampschulte, A.; Bruckschen, P.; Strauss, H. (2001-05-01). "The sulphur isotopic composition of trace sulphates in Carboniferous brachiopods: implications for coeval seawater, correlation with other geochemical cycles and isotope stratigraphy". Chemical Geology. Response of the Oceanic / Atmospheric Systems to Past Global Changes. 175 (1): 149–173. Bibcode:2001ChGeo.175..149K. doi:10.1016/S0009-2541(00)00367-3. ISSN 0009-2541.
- ↑ Busenberg, Eurybiades; Niel Plummer, L. (March 1985). "Kinetic and thermodynamic factors controlling the distribution of SO32− and Na+ in calcites and selected aragonites". Geochimica et Cosmochimica Acta. 49 (3): 713–725. Bibcode:1985GeCoA..49..713B. doi:10.1016/0016-7037(85)90166-8.
- ↑ Staudt, Wilfried J.; Schoonen, Martin A. A. (1995). "Sulfate Incorporation into Sedimentary Carbonates". Geochemical Transformations of Sedimentary Sulfur. ACS Symposium Series. Vol. 612. pp. 332–345. doi:10.1021/bk-1995-0612.ch018. ISBN 0-8412-3328-4.
- ↑ Claypool, George E.; Holser, William T.; Kaplan, Isaac R.; Sakai, Hitoshi; Zak, Israel (1980). "The age curves of sulfur and oxygen isotopes in marine sulfate and their mutual interpretation". Chemical Geology. 28: 199–260. Bibcode:1980ChGeo..28..199C. doi:10.1016/0009-2541(80)90047-9.
- ↑ Hardie, L. A. (1 March 1984). "Evaporites; marine or non-marine?". American Journal of Science. 284 (3): 193–240. Bibcode:1984AmJS..284..193H. doi:10.2475/ajs.284.3.193.
- ↑ Grover, G.; Harris, P. (1989). Subsurface and outcrop examination of the Capitan Shelf margin, northern Delaware Basin : SEPM Core Workshop no. 13, San Antonio, April 23, 1989. Tulsa, OK: Society of Economic Paleontologists and Mineralogists. ISBN 9780918985804.
- ↑ Wiley, N. (1989). Evolution of Global Biogeochemistry: Sulfur Cycle. pp. 57–64.
- ↑ Utrilla, Rosa; Pierre, Catherine; Orti, Federico; Pueyo, Juan José (December 1992). "Oxygen and sulphur isotope compositions as indicators of the origin of Mesozoic and Cenozoic evaporites from Spain". Chemical Geology. 102 (1–4): 229–244. Bibcode:1992ChGeo.102..229U. doi:10.1016/0009-2541(92)90158-2.
- ↑ Strauss, H. (August 1997). "The isotopic composition of sedimentary sulfur through time". Palaeogeography, Palaeoclimatology, Palaeoecology. 132 (1–4): 97–118. Bibcode:1997PPP...132...97S. doi:10.1016/S0031-0182(97)00067-9.
- ↑ Lu, F. H.; Meyers, W. J. (1 May 2003). "Sr, S, and OSO4 Isotopes and the Depositional Environments of the Upper Miocene Evaporites, Spain". Journal of Sedimentary Research. 73 (3): 444–450. Bibcode:2003JSedR..73..444L. doi:10.1306/093002730444.
- ↑ Playà, Elisabet; Cendón, Dioni I.; Travé, Anna; Chivas, Allan R.; García, Adriana (October 2007). "Non-marine evaporites with both inherited marine and continental signatures: The Gulf of Carpentaria, Australia, at ~70 ka". Sedimentary Geology. 201 (3–4): 267–285. Bibcode:2007SedG..201..267P. doi:10.1016/j.sedgeo.2007.05.010.
- ↑ Bishop, James K. B. (24 March 1988). "The barite-opal-organic carbonassociation inoceanic particulate matter". Nature. 332 (6162): 341–343. Bibcode:1988Natur.332..341B. doi:10.1038/332341a0. S2CID 4349970.
- ↑ Paytan, A.; Kastner, M.; Martin, E. E.; Macdougall, J. D.; Herbert, T. (2 December 1993). "Marine barite as a monitor of seawater strontium isotope composition". Nature. 366 (6454): 445–449. Bibcode:1993Natur.366..445P. doi:10.1038/366445a0. S2CID 4238837.
- ↑ Paytan, A. (20 November 1998). "Sulfur Isotopic Composition of Cenozoic Seawater Sulfate". Science. 282 (5393): 1459–1462. CiteSeerX 10.1.1.528.6626. doi:10.1126/science.282.5393.1459. PMID 9822370.
- ↑ Paytan, Adina; Mearon, Sarah; Cobb, Kim; Kastner, Miriam (2002). "Origin of marine barite deposits: Sr and S isotope characterization". Geology. 30 (8): 747. Bibcode:2002Geo....30..747P. doi:10.1130/0091-7613(2002)030<0747:OOMBDS>2.0.CO;2.
- ↑ Torres, M.E.; Brumsack, H.J.; Bohrmann, G.; Emeis, K.C. (January 1996). "Barite fronts in continental margin sediments: a new look at barium remobilization in the zone of sulfate reduction and formation of heavy barites in diagenetic fronts". Chemical Geology. 127 (1–3): 125–139. Bibcode:1996ChGeo.127..125T. doi:10.1016/0009-2541(95)00090-9.
- ↑ Meybeck, M. (2003). Global occurrence of major elements in rivers. Vol. 5. pp. 207–223. Bibcode:2003TrGeo...5..207M. doi:10.1016/B0-08-043751-6/05164-1. ISBN 9780080437514.
{{cite book}}:|journal=ignored (help) - ↑ Berner, E. K.; Berner, R. A. (2012). Global environment : water, air, and geochemical cycles (2nd ed.). Princeton, N.J.: Princeton University Press. ISBN 9780691136783.
- ↑ Ault, W.U; Kulp, J.L (July 1959). "Isotopic geochemistry of sulphur". Geochimica et Cosmochimica Acta. 16 (4): 201–235. Bibcode:1959GeCoA..16..201A. doi:10.1016/0016-7037(59)90112-7.
- ↑ Garrels, R. M.; Lerman, A. (1 November 1984). "Coupling of the sedimentary sulfur and carbon cycles; an improved model". American Journal of Science. 284 (9): 989–1007. Bibcode:1984AmJS..284..989G. doi:10.2475/ajs.284.9.989.
- ↑ Jarvis, I. (1995). "Phosphorite geochemistry:state-of-the-art and environmental concerns". Oceanographic Literature Review. 42 (8): 639.
- ↑ Alt, Jeffrey C. (1995). "Sulfur isotopic profile through the oceanic crust: Sulfur mobility and seawater-crustal sulfur exchange during hydrothermal alteration". Geology. 23 (7): 585. Bibcode:1995Geo....23..585A. doi:10.1130/0091-7613(1995)023<0585:SIPTTO>2.3.CO;2.
- 1 2 3 4 Canfield, D. E. (1 December 2004). "The evolution of the Earth surface sulfur reservoir". American Journal of Science. 304 (10): 839–861. Bibcode:2004AmJS..304..839C. doi:10.2475/ajs.304.10.839.
- ↑ Halevy, I.; Peters, S. E.; Fischer, W. W. (19 July 2012). "Sulfate Burial Constraints on the Phanerozoic Sulfur Cycle" (PDF). Science. 337 (6092): 331–334. Bibcode:2012Sci...337..331H. doi:10.1126/science.1220224. PMID 22822147. S2CID 25170268.
- ↑ Tostevin, Rosalie; Turchyn, Alexandra V.; Farquhar, James; Johnston, David T.; Eldridge, Daniel L.; Bishop, James K.B.; McIlvin, Matthew (June 2014). "Multiple sulfur isotope constraints on the modern sulfur cycle". Earth and Planetary Science Letters. 396: 14–21. Bibcode:2014E&PSL.396...14T. doi:10.1016/j.epsl.2014.03.057. hdl:1912/6760.
- ↑ Johnston, D. T.; Gill, B. C.; Masterson, A.; Beirne, E.; Casciotti, K. L.; Knapp, A. N.; Berelson, W. (2014-09-25). "Placing an upper limit on cryptic marine sulphur cycling". Nature. 513 (7519): 530–533. Bibcode:2014Natur.513..530J. doi:10.1038/nature13698. ISSN 1476-4687. PMID 25209667. S2CID 4469105.
- ↑ Rees, C.E.; Jenkins, W.J.; Monster, Jan (April 1978). "The sulphur isotopic composition of ocean water sulphate". Geochimica et Cosmochimica Acta. 42 (4): 377–381. Bibcode:1978GeCoA..42..377R. doi:10.1016/0016-7037(78)90268-5.
- ↑ Harrison, A. G.; Thode, H. G. (1958). "Mechanism of the bacterial reduction of sulphate from isotope fractionation studies". Transactions of the Faraday Society. 54: 84. doi:10.1039/TF9585400084.
- ↑ Habicht, Kirsten S.; Canfield, Donald E. (December 1997). "Sulfur isotope fractionation during bacterial sulfate reduction in organic-rich sediments". Geochimica et Cosmochimica Acta. 61 (24): 5351–5361. Bibcode:1997GeCoA..61.5351H. doi:10.1016/S0016-7037(97)00311-6. PMID 11541664.
- ↑ Canfield, D.E. (April 2001). "Isotope fractionation by natural populations of sulfate-reducing bacteria". Geochimica et Cosmochimica Acta. 65 (7): 1117–1124. Bibcode:2001GeCoA..65.1117C. doi:10.1016/S0016-7037(00)00584-6.
- ↑ Brunner, Benjamin; Bernasconi, Stefano M. (October 2005). "A revised isotope fractionation model for dissimilatory sulfate reduction in sulfate reducing bacteria". Geochimica et Cosmochimica Acta. 69 (20): 4759–4771. Bibcode:2005GeCoA..69.4759B. doi:10.1016/j.gca.2005.04.015.
- ↑ Sim, Min Sub; Ono, Shuhei; Donovan, Katie; Templer, Stefanie P.; Bosak, Tanja (August 2011). "Effect of electron donors on the fractionation of sulfur isotopes by a marine Desulfovibrio sp". Geochimica et Cosmochimica Acta. 75 (15): 4244–4259. Bibcode:2011GeCoA..75.4244S. doi:10.1016/j.gca.2011.05.021.
- 1 2 Jorgensen, B. B. (13 July 1990). "A Thiosulfate Shunt in the Sulfur Cycle of Marine Sediments". Science. 249 (4965): 152–154. Bibcode:1990Sci...249..152B. doi:10.1126/science.249.4965.152. PMID 17836966. S2CID 220093825.
- 1 2 Canfield, D.; Thamdrup, B (23 December 1994). "The production of 34S-depleted sulfide during bacterial disproportionation of elemental sulfur". Science. 266 (5193): 1973–1975. Bibcode:1994Sci...266.1973C. doi:10.1126/science.11540246. PMID 11540246.
- ↑ Jorgensen, Bo Barker (March 1979). "A theoretical model of the stable sulfur isotope distribution in marine sediments". Geochimica et Cosmochimica Acta. 43 (3): 363–374. Bibcode:1979GeCoA..43..363J. doi:10.1016/0016-7037(79)90201-1.
- ↑ Gomes, Maya L.; Hurtgen, Matthew T. (May 2015). "Sulfur isotope fractionation in modern euxinic systems: Implications for paleoenvironmental reconstructions of paired sulfate–sulfide isotope records". Geochimica et Cosmochimica Acta. 157: 39–55. Bibcode:2015GeCoA.157...39G. doi:10.1016/j.gca.2015.02.031.
- 1 2 Canfield, Donald E.; Teske, Andreas (11 July 1996). "Late Proterozoic rise in atmospheric oxygen concentration inferred from phylogenetic and sulphur-isotope studies". Nature. 382 (6587): 127–132. Bibcode:1996Natur.382..127C. doi:10.1038/382127a0. PMID 11536736. S2CID 4360682.
- 1 2 Canfield, D.; Raiswell, R. (1999). "The evolution of the sulfur cycle". American Journal of Science. 299 (7–9): 697–723. Bibcode:1999AmJS..299..697C. doi:10.2475/ajs.299.7-9.697. S2CID 5354992.
- ↑ Gill, Benjamin C.; Lyons, Timothy W.; Frank, Tracy D. (2008-10-01). "Behavior of carbonate-associated sulfate during meteoric diagenesis and implications for the sulfur isotope paleoproxy". Geochimica et Cosmochimica Acta. 72 (19): 4699–4711. Bibcode:2008GeCoA..72.4699G. doi:10.1016/j.gca.2008.07.001. ISSN 0016-7037.
- ↑ Takano, B. (June 1985). "Geochemical implications of sulfate in sedimentary carbonates". Chemical Geology. 49 (4): 393–403. Bibcode:1985ChGeo..49..393T. doi:10.1016/0009-2541(85)90001-4.
- ↑ Pingitore, Nicholas E.; Meitzner, George; Love, Karen M. (June 1995). "Identification of sulfate in natural carbonates by x-ray absorption spectroscopy". Geochimica et Cosmochimica Acta. 59 (12): 2477–2483. Bibcode:1995GeCoA..59.2477P. doi:10.1016/0016-7037(95)00142-5.
- ↑ Kontrec, Jasminka; Kralj, Damir; Bre?evi?, Ljerka; Falini, Giuseppe; Fermani, Simona; Noethig-Laslo, Vesna; Mirosavljevi?, Krunoslav (December 2004). "Incorporation of Inorganic Anions in Calcite". European Journal of Inorganic Chemistry. 2004 (23): 4579–4585. doi:10.1002/ejic.200400268.
- ↑ Fernández-Díaz, Lurdes; Fernández-González, Ángeles; Prieto, Manuel (November 2010). "The role of sulfate groups in controlling CaCO3 polymorphism" (PDF). Geochimica et Cosmochimica Acta. 74 (21): 6064–6076. Bibcode:2010GeCoA..74.6064F. doi:10.1016/j.gca.2010.08.010. hdl:10651/10897.
- ↑ Balan, Etienne; Blanchard, Marc; Pinilla, Carlos; Lazzeri, Michele (May 2014). "First-principles modeling of sulfate incorporation and 34S/32S isotopic fractionation in different calcium carbonates". Chemical Geology. 374–375: 84–91. Bibcode:2014ChGeo.374...84B. doi:10.1016/j.chemgeo.2014.03.004.
- 1 2 3 Paris, G.; Adkins, J. F.; Sessions, A. L.; Webb, S. M.; Fischer, W. W. (6 November 2014). "Neoarchean carbonate-associated sulfate records positive 33S anomalies". Science. 346 (6210): 739–741. doi:10.1126/science.1258211. PMID 25378622. S2CID 20532947.
- ↑ de Groot, Pier A. (2009). Handbook of stable isotope analytical techniques (1st ed.). Amsterdam: Elsevier. ISBN 9780444511157.
- ↑ Owens, Jeremy D.; Gill, Benjamin C.; Jenkyns, Hugh C.; Bates, Steven M.; Severmann, Silke; Kuypers, Marcel M. M.; Woodfine, Richard G.; Lyons, Timothy W. (2013-11-12). "Sulfur isotopes track the global extent and dynamics of euxinia during Cretaceous Oceanic Anoxic Event 2". Proceedings of the National Academy of Sciences of the United States of America. 110 (46): 18407–18412. Bibcode:2013PNAS..11018407O. doi:10.1073/pnas.1305304110. ISSN 1091-6490. PMC 3831968. PMID 24170863.
- ↑ Gill, Benjamin C.; Lyons, Timothy W.; Young, Seth A.; Kump, Lee R.; Knoll, Andrew H.; Saltzman, Matthew R. (2011-01-06). "Geochemical evidence for widespread euxinia in the later Cambrian ocean". Nature. 469 (7328): 80–83. Bibcode:2011Natur.469...80G. doi:10.1038/nature09700. ISSN 1476-4687. PMID 21209662. S2CID 4319979.
- ↑ Gill, Benjamin C.; Lyons, Timothy W.; Jenkyns, Hugh C. (2011-12-15). "A global perturbation to the sulfur cycle during the Toarcian Oceanic Anoxic Event". Earth and Planetary Science Letters. 312 (3): 484–496. Bibcode:2011E&PSL.312..484G. doi:10.1016/j.epsl.2011.10.030. ISSN 0012-821X.
- ↑ Kampschulte, A.; Bruckschen, P.; Strauss, H. (May 2001). "The sulphur isotopic composition of trace sulphates in Carboniferous brachiopods: implications for coeval seawater, correlation with other geochemical cycles and isotope stratigraphy". Chemical Geology. 175 (1–2): 149–173. Bibcode:2001ChGeo.175..149K. doi:10.1016/S0009-2541(00)00367-3.
- ↑ Marenco, Pedro J.; Corsetti, Frank A.; Hammond, Douglas E.; Kaufman, Alan J.; Bottjer, David J. (January 2008). "Oxidation of pyrite during extraction of carbonate associated sulfate". Chemical Geology. 247 (1–2): 124–132. Bibcode:2008ChGeo.247..124M. doi:10.1016/j.chemgeo.2007.10.006.
- ↑ Wotte, Thomas; Shields-Zhou, Graham A.; Strauss, Harald (October 2012). "Carbonate-associated sulfate: Experimental comparisons of common extraction methods and recommendations toward a standard analytical protocol". Chemical Geology. 326–327: 132–144. Bibcode:2012ChGeo.326..132W. doi:10.1016/j.chemgeo.2012.07.020.
- ↑ Theiling, Bethany P.; Coleman, Max (September 2015). "Refining the extraction methodology of carbonate associated sulfate: Evidence from synthetic and natural carbonate samples". Chemical Geology. 411: 36–48. Bibcode:2015ChGeo.411...36T. doi:10.1016/j.chemgeo.2015.06.018.
- ↑ Crowe, S. A.; Paris, G.; Katsev, S.; Jones, C.; Kim, S.-T.; Zerkle, A. L.; Nomosatryo, S.; Fowle, D. A.; Adkins, J. F.; Sessions, A. L.; Farquhar, J.; Canfield, D. E. (6 November 2014). "Sulfate was a trace constituent of Archean seawater" (PDF). Science. 346 (6210): 735–739. Bibcode:2014Sci...346..735C. doi:10.1126/science.1258966. PMID 25378621. S2CID 206561027.
- ↑ Shen, Yanan; Buick, Roger; Canfield, Donald E. (1 March 2001). "Isotopic evidence for microbial sulphate reduction in the early Archaean era". Nature. 410 (6824): 77–81. Bibcode:2001Natur.410...77S. doi:10.1038/35065071. PMID 11242044. S2CID 25375808.
- ↑ Hurtgen, Matthew T.; Arthur, Michael A.; Halverson, Galen P. (2005). "Neoproterozoic sulfur isotopes, the evolution of microbial sulfur species, and the burial efficiency of sulfide as sedimentary pyrite". Geology. 33 (1): 41. Bibcode:2005Geo....33...41H. doi:10.1130/G20923.1.