Asymmetric ester hydrolysis with pig liver esterase is the enantioselective conversion of an ester to a carboxylic acid through the action of the enzyme pig liver esterase (EC 3.1.1.1). Asymmetric ester hydrolysis involves the selective reaction of one of a pair of either enantiotopic (within the same molecule and related by a symmetry plane of the molecule) or enantiomorphic (in enantiomeric molecules and related as mirror images) ester groups.[1]
Introduction
Enzymes, which are composed of chiral amino acids, catalyze chemical reactions with high stereoselectivity. Specifically, esterase enzymes catalyze the hydrolysis of esters to carboxylic acids. This transformation may be rendered asymmetric if two enantiotopic ester groups exist in the substrate or if a racemic mixture of chiral esters is used. In the former case (desymmetrization), the chiral environment of the enzyme active site leads to selective hydrolysis of the ester that is closer to the catalytically active serine residue when the substrate is bound to the enzyme. In the latter case (kinetic resolution), one of the enantiomers is hydrolyzed faster than the other, leading to an excess of hydrolyzed product from one enantiomer. Both strategies rely on the fact that the transition states for hydrolysis of enantiotopic or enantiomorphic ester groups by the chiral enzyme are diastereomeric.[2]
Pig liver esterase (PLE) is a widely used enzyme for asymmetric ester hydrolysis. Although it was originally used for the desymmetrizing hydrolysis of glutarate esters,[3] PLE also hydrolyzes malonates, cyclic diesters, monoesters, and other substrates. Active site models have been advanced to explain the selectivity of PLE.[4]
(1)

Mechanism and Stereochemistry
Prevailing Mechanism
The active site of PLE facilitates both substrate binding and hydrolysis. A key serine residue in the active site promotes hydrolysis, but the substrate must present an ester group to this residue after binding to the enzyme active site for hydrolysis to take place. Whether the substrate is able to present an ester group to the catalytic serine residue depends on its bound conformation in the active site, which is dictated by amino acid side-chains in the active site. Thus, active site models of PLE have been advanced with the goal of predicting from the structure of the substrate which of two enantiotopic ester groups will be hydrolyzed (or whether hydrolysis is likely to occur at all).
A simple model for the binding conformation of an ester in the active site of PLE is shown below. This model accurately predicts the configuration of hydrolyzed glutarates and similar substrates.
(2)

Scope and Limitations
Although the substrate scope of PLE is broad, enantioselectivity varies as a function of the structure of the substrate. This section describes substrates that are hydrolyzed by PLE with the highest enantioselectivity, as well as sensitive substrates that may be hydrolyzed to achiral carboxylic acids in high yield without side reactions.
Glutarates were the first substrates to be hydrolyzed with PLE in high enantioselectivity. Although yields are moderate, enantioselectivity is extremely high.[5]
(3)

3-Alkyl glutarates with small alkyl substituents are hydrolyzed to the (R)-monoester; however, when a large alkyl substituent is present, the (S)-monoester forms.[6] This switch in enantioselectivity is accurately predicted by the active site model given above.
(4)

An opposite trend is observed in desymmetrizing hydrolyses of 2-methyl malonates, which afford the (S) enantiomer when the other substituent on C-2 is small, and the (R) enantiomer when the other C-2 substituent is large.[7]
(5)
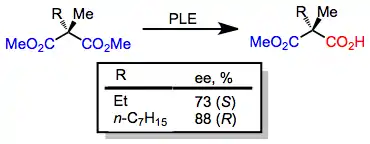
A number of meso diesters other than the substrates described above may be hydrolyzed by PLE with high enantioselectivity. Cyclic meso diesters tend to be hydrolyzed more selectively than acyclic diesters.[8] The predominant enantiomer of product depends on ring size.[9][10]
(6)

7-Oxabicyclo[2.2.1]heptane-2,3-dicarboxylates are an interesting class of diesters that are hydrolyzed by PLE with high enantioselectivity.[11] These substrates have been used for the enantioselective construction of biologically relevant sugars (see Synthetic Applications below).
(7)

Racemic mixtures of all of the substrates described above, as well as additional chiral diesters (such as the epoxy ester in equation (8)), may be resolved using PLE for kinetic resolution.[12] A significant disadvantage of kinetic resolution is a maximum yield of hydrolyzed product of 50%. However, if rapid racemization is occurring alongside hydrolysis (an example of dynamic kinetic resolution), a maximum yield of 100% is possible.[13]
(8)

Esterase enzymes may also be used for hydrolysis of base-sensitive monoesters. PLE has been applied to the synthesis of prostaglandins for the selective hydrolysis of the ester without destruction of the β-hydroxy ketone moiety.[14]
(9)

Synthetic Applications
A number of synthetic targets possess hidden symmetry that may be discovered by applying a retrosynthetic "symmetrizing" transform. In the forward direction, this operation corresponds to a desymmetrization reaction. For instance, mevalonolactone may be synthesized rapidly from a symmetric diester via desymmetrizing hydrolysis, chemoselective reduction, and lactonization.[5] Although the product itself is asymmetric, desymmetrization and functional group manipulations permit its synthesis from an achiral starting material.
(10)

Enantioselective hydrolysis of a conjugated diester followed by ozonolysis affords the skeleton of ribose. The resulting sugars are then carried on for the synthesis of nucleosides.[15]
(11)

L-α-Methyldopa may be rapidly synthesized from an achiral malonate through a sequence beginning with desymmetrization. Subsequent chemoselective transformations convert the carboxylic acid to an amine.[16]
(12)

Comparison with Other Methods
Other enzymes that may be used for asymmetric ester hydrolysis include electric eel acetylcholinesterase,[17] chymotrypsin,[3] and Baker's yeast.[18] The substrate scope of these enzymes differs from PLE, and in some cases they may provide hydrolyzed products in higher yield or enantioselectivity than PLE. Microorganisms may also be used for enantioselective hydrolysis;[19] however, difficulties associated with the handling of microorganisms have made these methods unpopular for organic synthesis.
Nonenzymatic methods for the differentiation of enantiotopic groups employ chiral catalysts or auxiliaries. For instance, the introduction of a chiral leaving group on both carboxylic acid groups of a meso diacid leads to selective attack by an achiral nucleophile at one of the (now) diastereotopic carbonyl groups.[20]
(13)

Experimental Conditions and Procedure
Typical Conditions
Enzymatic reactions are limited by the need for aqueous solvent and near-neutral reaction conditions. PLE hydrolyses are typically carried out with a phosphate buffer to maintain the pH between 7 and 8. As solubility of the substrate in the aqueous medium is critical, a small amount of a polar organic co-solvent is sometimes added to the aqueous solution of the enzyme. Commercially available PLE is of sufficient purity for most applications.
References
- ↑ Ohno, M.; Otsuka, M. Org. React. 1989, 37, 1. doi:10.1002/0471264180.or037.01
- ↑ Rétey, J.; Robinson, J. Stereospecificity in Organic Chemistry and Enzymology, Verlag Chemie, Weinheim, 1982.
- 1 2 Cohen, S.; Khedouri, E. J. Am. Chem. Soc. 1961, 83, 1093.
- ↑ Zemlicka, J.; Craine, L.; Heeg, M.-J.; Oliver, J. J. Org. Chem. 1988, 53, 937.
- 1 2 Huang, F.-C.; Lee, L. F. H.; Mittal, R. S. D.; Ravikumar, P. R.; Chan, J. A.; Sih, C. J.; Capsi, E.; Eck, C. R. J. Am. Chem. Soc. 1975, 97, 4144.
- ↑ Lam, L. K. P.; Hui, R. A. H. F.; Jones, J. B. J. Org. Chem. 1986, 51, 2047.
- ↑ Björkling, F.; Boutelje, J.; Gatenbeck, S.; Hult, K.; Norin, T.; Szmulik, P. Tetrahedron 1985, 41, 1347.
- ↑ Mohr, P.; Waespe-Sarevi, N.; Tamm, C.; Gawronska, K.; Gawronski, J. Helv. Chim. Acta 1983, 66, 2501.
- ↑ Sabbioni, G.; Shea, M. L.; Jones, J. B. J. Chem. Soc., Chem. Commun. 1984, 236.
- ↑ Schneider, M.; Engel, N.; Hönicke, P.; Heinemann, G.; Görisch, H. Angew. Chem. Int. Ed. Engl. 1984, 23, 67.
- ↑ Guanti, G.; Banfi, L.; Narisano, E.; Riva, R.; Thea, S. Tetrahedron Lett. 1986, 27, 4639.
- ↑ Mohr, P.; Rösslein, L.; Tamm, C. Helv. Chim. Acta 1987, 70, 142.
- ↑ Allen, J.; Williams, J. Tetrahedron Lett. 1996, 37, 1859.
- ↑ Hazato, A.; Tanaka, T.; Toru, T.; Okamura, N.; Bannai, K.; Sugiura, S.; Manabe, K.; Kurozumi, S. Nippon Kagaku Kaishi 1983, 9, 1390 [C.A., 100, 120720q (1984)].
- ↑ Ohno, M.; Kobayashi, S.; Adachi, K. in Enzymes as Catalysts in Organic Synthesis, Schneider, M. P. Ed., D. Reidel Publishing, Dordrecht, 1986, pp. 123–142.
- ↑ Björkling, F.; Boutelje, J.; Gatenbeck, S.; Hult, K.; Norin, T. Tetrahedron Lett. 1985, 26, 4957.
- ↑ Deardorff, D. R.; Matthews, A. J.; McMeekin, D. S.; Craney, C. L. Tetrahedron Lett. 1986, 27, 1255.
- ↑ Kerscher, V.; Kreiser, W. Tetrahedron Lett. 1987, 28, 531.
- ↑ Kotani, H.; Kuze, Y.; Uchida, S.; Miyabe, T.; Iimori, T.; Okano, K.; Kobayashi, S.; Ohno, M.; Agric. Biol. Chem. 1983, 47, 1363.
- ↑ Nagao, Y.; Ikeda, T.; Yagi, M.; Fujita, E.; Shiro, M. J. Am. Chem. Soc. 1982, 104, 2079.