| MEN type 2A (Sipple syndrome) | |
|---|---|
| Other names | MEN2 |
 | |
| Bilateral pheochromocytomas associated with Multiple endocrine neoplasia type 2 | |
| Specialty | Oncology |
Multiple endocrine neoplasia type 2 (also known as "Pheochromocytoma (codons 630 and 634) and amyloid producing medullary thyroid carcinoma",[1] "PTC syndrome,"[1] and "Sipple syndrome"[1]) is a group of medical disorders associated with tumors of the endocrine system. The tumors may be benign or malignant (cancer). They generally occur in endocrine organs (e.g. thyroid, parathyroid, and adrenals), but may also occur in endocrine tissues of organs not classically thought of as endocrine.[2] MEN2 is a sub-type of MEN (multiple endocrine neoplasia) and itself has sub-types, as discussed below. Variants in MEN2A have been associated with Hirschsprung disease. Screening for this condition can begin as young as eight years old for Pheochromocytoma.
Signs and symptoms
MEN2 can present with a sign or symptom related to a tumor or, in the case of multiple endocrine neoplasia type 2b, with characteristic musculoskeletal and/or lip and/or gastrointestinal findings. Medullary thyroid carcinoma (MTC) represents the most frequent initial diagnosis. Occasionally pheochromocytoma or primary hyperparathyroidism may be the initial diagnosis.
Pheochromocytoma occurs in 33-50% of MEN2 cases.[3] In MEN2A, primary hyperparathyroidism occurs in 10–50% of cases and is usually diagnosed after the third decade of life. Rarely, it may present in childhood or be the sole clinical manifestation of this syndrome.MEN2A associates medullary thyroid carcinoma with pheochromocytoma in about 20–50% of cases and with primary hyperparathyroidism in 5–20% of cases. MEN2B associates medullary thyroid carcinoma with pheochromocytoma in 50% of cases, with marfanoid habitus and with mucosal and digestive neurofibromatosis.
In familial isolated medullary thyroid carcinoma the other components of the disease are absent. In a review of 85 patients 70 had MEN2A and 15 had MEN2B.[3] The initial manifestation of MEN2 was medullary thyroid carcinoma in 60% of patients, medullary thyroid carcinoma synchronous with pheochromocytoma in 34% and pheochromocytoma alone in 6%. 72% had bilateral pheochromocytomas.
Causes
The table in the multiple endocrine neoplasia article lists the genes involved in the various MEN syndromes. Most cases of MEN2 derive from a variation in the RET proto-oncogene, and are specific for cells of neural crest origin. A database of MEN-implicated RET mutations is maintained by the University of Utah Department of Physiology.[4]
The protein produced by the RET gene plays an important role in the TGF-beta (transforming growth factor beta) signaling system. Because the TGF-beta system operates in nervous tissues throughout the body, variations in the RET gene can have effects in nervous tissues throughout the body. MEN2 generally results from a gain-of-function variant of a RET gene. Other diseases, such as Hirschsprung disease, result from loss-of-function variants. OMIM # 164761 lists the syndromes associated with the RET gene.
Genetics

When inherited, multiple endocrine neoplasia type 2 is transmitted in an autosomal dominant pattern, which means affected people have one affected parent, and possibly affected siblings and children. Some cases, however, result from spontaneous new mutations in the RET gene. These cases occur in people with no family history of the disorder. In MEN2B, for example, about half of all cases arise as spontaneous new mutations.
Diagnosis
Diagnosis is suspected when a patient with family history of two of the three classical tumors (medullary thyroid cancer, pheochromocytoma, parathyroid adenoma) or MEN2 presents with one of the classical tumors. It is confirmed by genetic testing for mutation in RET gene.[5]
Differences in presentation
As noted, all types of MEN2 include pheochromocytoma and medullary thyroid carcinoma.
MEN2A is additionally characterized by the presence of parathyroid hyperplasia.
MEN2B is additionally characterized by the presence of mucocutaneous neuroma, gastrointestinal symptoms (e.g. constipation and flatulence), and muscular hypotonia.
Classification
Before gene testing was available, the type and location of tumors determined which type of MEN2 a person had. Gene testing now allows a diagnosis before tumors or symptoms develop.
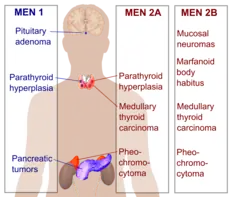
A table in the multiple endocrine neoplasia article compares the various MEN syndromes. MEN2 and MEN1 are distinct conditions, despite their similar names. MEN2 includes MEN2A, MEN2B and familial medullary thyroid cancer (FMTC).
The common feature among the three sub-types of MEN2 is a high propensity to develop medullary thyroid carcinoma.
A variant of MEAs 2A was described in 1989.[7] This variant also has patches of cutaneous amyloidosis in the mid/upper back and is inherited in an autosomal dominant fashion.[8]
Management
Management of MEN2 patients includes thyroidectomy including cervical central and bilateral lymph nodes dissection for MTC, unilateral adrenalectomy for unilateral pheochromocytoma or bilateral adrenalectomy when both glands are involved and selective resection of pathologic parathyroid glands for primary hyperparathyroidism. Familial genetic screening is recommended to identify at risk subjects who will develop the disease, permitting early management by performing prophylactic thyroidectomy, giving them the best chance of cure.
Prognosis
Prognosis of MEN2 is mainly related to the stage-dependant prognosis of MTC indicating the necessity of a complete thyroid surgery for index cases with MTC and the early thyroidectomy for screened at risk subjects.
See also
References
- 1 2 3 Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ↑ Moline J, Eng C (2011). "Multiple endocrine neoplasia type 2: an overview". Genet. Med. 13 (9): 755–64. doi:10.1097/GIM.0b013e318216cc6d. PMID 21552134. S2CID 22402472.
- 1 2 Thosani S, Ayala-Ramirez M, Palmer L, Hu MI, Rich T, Gagel RF, Cote G, Waguespack SG, Habra MA, Jimenez C (2013) The characterization of pheochromocytoma and its impact on overall survival in Multiple Endocrine Neoplasia type 2. J Clin Endocrinol Metab
- ↑ "MEN2 Database". University of Utah.
- ↑ "Multiple Endocrine Neoplasia Type 2". Cancer.Net. 2012-06-25. Retrieved 2018-01-15.
- ↑ Wray CJ, Rich TA, Waguespack SG, Lee JE, Perrier ND, Evans DB (January 2008). "Failure to recognize multiple endocrine neoplasia 2B: more common than we think?". Ann. Surg. Oncol. 15 (1): 293–301. doi:10.1245/s10434-007-9665-4. PMID 17963006. S2CID 2564555.
- ↑ Donovan DT, Levy ML, Furst EJ, Alford BR, Wheeler T, Tschen JA, Gagel RF (1989) Familial cutaneous lichen amyloidosis in association with multiple endocrine neoplasia type 2A: a new variant. Henry Ford Hosp Med J 37(3-4):147-150
- ↑ Jabbour SA, Davidovici BB, Wolf R (2006) Rare syndromes. Clin Dermatol 24(4):299-316