
The SOS response is a global response to DNA damage in which the cell cycle is arrested and DNA repair and mutagenesis is induced. The system involves the RecA protein (Rad51 in eukaryotes). The RecA protein, stimulated by single-stranded DNA, is involved in the inactivation of the repressor (LexA) of SOS response genes thereby inducing the response. It is an error-prone repair system that contributes significantly to DNA changes observed in a wide range of species.
Discovery
The SOS response was articulated by Evelyn Witkin.[3][4] Later, by characterizing the phenotypes of mutagenised E. coli, she and post doctoral student Miroslav Radman detailed the SOS response to UV radiation in bacteria.[3][5] The SOS response to DNA damage was a seminal discovery because it was the first coordinated stress response to be elucidated.[6]
Mechanism
During normal growth, the SOS genes are negatively regulated by LexA repressor protein dimers. Under normal conditions, LexA binds to a 20-bp consensus sequence (the SOS box) in the operator region for those genes. Some of these SOS genes are expressed at certain levels even in the repressed state, according to the affinity of LexA for their SOS box. Activation of the SOS genes occurs after DNA damage by the accumulation of single stranded (ssDNA) regions generated at replication forks, where DNA polymerase is blocked. RecA forms a filament around these ssDNA regions in an ATP-dependent fashion, and becomes activated.[7] The activated form of RecA interacts with the LexA repressor to facilitate the LexA repressor's self-cleavage from the operator.[7][8]
Once the pool of LexA decreases, repression of the SOS genes goes down according to the level of LexA affinity for the SOS boxes.[7] Operators that bind LexA weakly are the first to be fully expressed. In this way LexA can sequentially activate different mechanisms of repair. Genes having a weak SOSbox (such as lexA, recA, uvrA, uvrB, and uvrD) are fully induced in response to even weak SOS-inducing treatments. Thus the first SOS repair mechanism to be induced is nucleotide excision repair (NER), whose aim is to fix DNA damage without commitment to a full-fledged SOS response. If, however, NER does not suffice to fix the damage, the LexA concentration is further reduced, so the expression of genes with stronger LexA boxes (such as sulA, umuD, umuC – these are expressed late) is induced.[7] SulA stops cell division[7] by binding to FtsZ, the initiating protein in this process. This causes filamentation, and the induction of UmuDC-dependent mutagenic repair. As a result of these properties, some genes may be partially induced in response to even endogenous levels of DNA damage, while other genes appear to be induced only when high or persistent DNA damage is present in the cell.
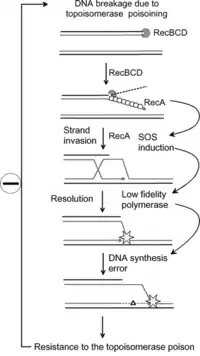
Antibiotic resistance
Research has shown that the SOS response system can lead to mutations which can lead to resistance to antibiotics.[9] The increased rate of mutation during the SOS response is caused by three low-fidelity DNA polymerases: Pol II, Pol IV and Pol V.[10][9] Researchers are now targeting these proteins with the aim of creating drugs that prevent SOS repair. By doing so, the time needed for pathogenic bacteria to evolve antibiotic resistance could be extended, thus improving the long term viability of some antibiotic drugs.[11]
As well as genetic resistance the SOS response can also promote phenotypic resistance. Here, the genome is preserved whilst other non-genetic factors are altered to enable the bacteria to survive. The SOS dependent tisB-istR toxin-antitoxin system has, for example, been linked to DNA damage-dependent persister cell induction.[12]
Genotoxicity testing
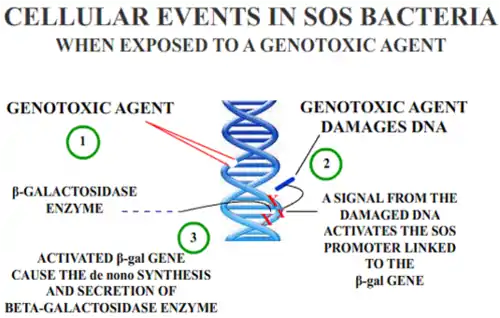
In Escherichia coli, different classes of DNA-damaging agents can initiate the SOS response, as described above. Taking advantage of an operon fusion placing the lac operon (responsible for producing beta-galactosidase, a protein which degrades lactose) under the control of an SOS-related protein, a simple colorimetric assay for genotoxicity is possible. A lactose analog is added to the bacteria, which is then degraded by beta-galactosidase, thereby producing a colored compound which can be measured quantitatively through spectrophotometry. The degree of color development is an indirect measure of the beta-galactosidase produced, which itself is directly related to the amount of DNA damage.
The E. coli are further modified in order to have a number of mutations including a uvrA mutation which renders the strain deficient in excision repair, increasing the response to certain DNA-damaging agents, as well as an rfa mutation, which renders the bacteria lipopolysaccharide-deficient, allowing better diffusion of certain chemicals into the cell in order to induce the SOS response.[13] Commercial kits which measures the primary response of the E. coli cell to genetic damage are available and may be highly correlated with the Ames Test for certain materials.[14]
Additional images
 The SOS response inhibits septum formation until bacterial DNA can be repaired and is observable as filamentation when cells are examined by microscopy (top right of image).
The SOS response inhibits septum formation until bacterial DNA can be repaired and is observable as filamentation when cells are examined by microscopy (top right of image).
See also
References
- ↑ Little JW, Mount DW (May 1982). "The SOS regulatory system of Escherichia coli". Cell. 29 (1): 11–22. doi:10.1016/0092-8674(82)90085-X. PMID 7049397. S2CID 12476812.
- ↑ Michel B (July 2005). "After 30 years of study, the bacterial SOS response still surprises us". PLOS Biology. 3 (7): e255. doi:10.1371/journal.pbio.0030255. PMC 1174825. PMID 16000023.

- 1 2 Fitzgerald, Devon M.; Hastings, P.J.; Rosenberg, Susan M. (6 March 2017). "Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance". Annual Review of Cancer Biology. 1 (1): 119–140. doi:10.1146/annurev-cancerbio-050216-121919. ISSN 2472-3428. PMC 5794033. PMID 29399660. Retrieved 6 March 2023.
- ↑ Witkin, E M (May 1967). "The radiation sensitivity of Escherichia coli B: a hypothesis relating filament formation and prophage induction". Proceedings of the National Academy of Sciences. 57 (5): 1275–1279. Bibcode:1967PNAS...57.1275W. doi:10.1073/pnas.57.5.1275. ISSN 0027-8424. PMC 224468. PMID 5341236.
- ↑ Witkin, E. M. (1976). "Ultraviolet mutagenesis and inducible DNA repair in Escherichia coli". Bacteriological Reviews. 40 (4): 869–907. doi:10.1128/MMBR.40.4.869-907.1976. PMC 413988. PMID 795416.
- ↑ Radman, M (1975). "Phenomenology of an inducible mutagenic DNA repair pathway in Escherichia coli: SOS repair hypothesis". Basic Life Sciences. 5A: 355–367. doi:10.1007/978-1-4684-2895-7_48. PMID 1103845.
- 1 2 3 4 5 Maslowska, K. H.; Makiela-Dzbenska, K.; Fijalkowska, I. J. (May 2019). "The SOS system: A complex and tightly regulated response to DNA damage". Environmental and Molecular Mutagenesis. 60 (4): 368–384. Bibcode:2019EnvMM..60..368M. doi:10.1002/em.22267. PMC 6590174. PMID 30447030.
- ↑ Nelson DL, Cox MM (April 2004) Lehninger Principles of Biochemistry 4th Edition. New York: W.H. Freeman and Company. page 1098.
- 1 2 Cirz, RT; Chin, JK; Andes, DR; De Crécy-Lagard, V; Craig, WA; Romesberg, FE; et al. (June 2005). "Inhibition of mutation and combating the evolution of antibiotic resistance". PLOS Biology. 3 (6): e176. doi:10.1371/journal.pbio.0030176. PMC 1088971. PMID 15869329.
- ↑ Jaszczur, M; Bertram, JG; Robinson, A; van Oijen, AM; Woodgate, R; Cox, MM; Goodman, MF (April 2016). "Mutations for Worse or Better: Low Fidelity DNA Synthesis by SOS DNA Polymerase V is a Tightly-Regulated Double-Edged Sword". Biochemistry. 55 (16): 2309–2318. doi:10.1021/acs.biochem.6b00117. PMC 4846499. PMID 27043933.
- ↑ Lee, AM; Ross, CT; Zeng, BB; Singleton, SF; et al. (July 2005). "A molecular target for suppression of the evolution of antibiotic resistance: Inhibition of the Escherichia coli RecA Protein by N6-(1-Naphthyl)-ADP". Journal of Medicinal Chemistry. 48 (17): 5408–5411. doi:10.1021/jm050113z. PMID 16107138.
- ↑ Dörr, T; Vulić, M; Lewis, K (February 2010). "Ciprofloxacin Causes Persister Formation by Inducing the TisB toxin in Escherichia coli". PLOS Biology. 8 (2): e1000317. doi:10.1371/journal.pbio.1000317. PMC 2826370. PMID 20186264.
- ↑ Quillardet P, Hofnung M (October 1993). "The SOS chromotest: A review". Mutation Research. 297 (3): 235–279. doi:10.1016/0165-1110(93)90019-J. PMID 7692273.
- ↑ Quillardet P, Hofnung M (June 1985). "The SOS chromotest, a colorimetric bacterial assay for genotoxins: validation study with 83 compounds". Mutation Research. 147 (3): 79–95. doi:10.1016/0165-1161(85)90021-4. PMID 3923333.
External links
| Excision repair | |
|---|---|
| Other forms of repair | |
| Other/ungrouped proteins | |
| Regulation | |
| Other/ungrouped | |
| |