Progeroid syndromes (PS) are a group of rare genetic disorders that mimic physiological aging, making affected individuals appear to be older than they are.[1][2] The term progeroid syndrome does not necessarily imply progeria (Hutchinson–Gilford progeria syndrome), which is a specific type of progeroid syndrome.
Progeroid means "resembling premature aging", a definition that can apply to a broad range of diseases. Familial Alzheimer's disease and familial Parkinson's disease are two well-known accelerated-aging diseases that are more frequent in older individuals. They affect only one tissue and can be classified as unimodal progeroid syndromes. Segmental progeria, which is more frequently associated with the term progeroid syndrome, tends to affect multiple or all tissues while causing affected individuals to exhibit only some of the features associated with aging.
All disorders within this group are thought to be monogenic,[3] meaning they arise from mutations of a single gene. Most known PS are due to genetic mutations that lead to either defects in the DNA repair mechanism or defects in lamin A/C.
Examples of PS include Werner syndrome (WS), Bloom syndrome (BS), Rothmund–Thomson syndrome (RTS), Cockayne syndrome (CS), xeroderma pigmentosum (XP), trichothiodystrophy (TTD), combined xeroderma pigmentosum-Cockayne syndrome (XP-CS), restrictive dermopathy (RD), and Hutchinson–Gilford progeria syndrome (HGPS). Individuals with these disorders tend to have a reduced lifespan.[3] Progeroid syndromes have been widely studied in the fields of aging, regeneration, stem cells, and cancer. The most widely studied of the progeroid syndromes are Werner syndrome and Hutchinson–Gilford progeria, as they are seen to most resemble natural aging.[3]
Defects in DNA repair
One of the main causes of progeroid syndromes is genetic mutations, which lead to defects in the cellular processes which repair DNA. The DNA damage theory of aging proposes that aging is a consequence of the accumulation of naturally occurring DNA damages. The accumulated damage may arise from reactive oxygen species (ROS), chemical reactions (e.g. with intercalating agents), radiation, depurination, and deamination.
Mutations in three classes of DNA repair proteins, RecQ protein-like helicases (RECQLs), nucleotide excision repair (NER) proteins, and nuclear envelope proteins LMNA (lamins) have been associated with the following progeroid syndromes:
- Werner syndrome (WS)
- Bloom syndrome (BS)
- Rothmund–Thomson syndrome (RTS)
- Cockayne syndrome (CS)
- Xeroderma pigmentosum (XP)
- Trichothiodystrophy (TTD)
RecQ-associated PS
RecQ is a family of conserved ATP-dependent helicases required for repairing DNA and preventing deleterious recombination and genomic instability.[4] DNA helicases are enzymes that bind to double-stranded DNA and temporarily separate them. This unwinding is required in replication of the genome during mitosis, but in the context of PS, it is a required step in repairing damaged DNA. Thus, DNA helicases, such as RecQ, maintain the integrity of a cell, and defects in these helicases are linked to an increased predisposition to cancer and aging phenotypes.[5] Thus, individuals with RecQ-associated PS show an increased risk of developing cancer,[6] which is caused by genomic instability and increased rates of mutation.[7]
There are five genes encoding RecQ in humans (RECQ1-5), and defects in RECQL2/WRN, RECQL3/BLM and RECQL4 lead to Werner syndrome (WS), Bloom syndrome (BS), and Rothmund–Thomson syndrome (RTS), respectively.[4][8] On the cellular level, cells of affected individuals exhibit chromosomal abnormalities, genomic instability, and sensitivity to mutagens.[7]
Werner syndrome

Werner syndrome (WS) is a rare autosomal recessive disorder.[9][10] It has a global incidence rate of less than 1 in 100,000 live births,[9] although incidences in Japan and Sardinia are higher, where it affects 1 in 20,000-40,000 and 1 in 50,000, respectively.[11][12] As of 2006, there were approximately 1,300 reported cases of WS worldwide.[3] Affected individuals typically grow and develop normally until puberty, when they do not experience the typical adolescent growth spurt. The mean age of diagnosis is twenty-four.[13] The median and mean age of death are 47-48 and 54 years, respectively;[14] the main cause of death is cardiovascular disease or cancer.[3][13]
Affected individuals can exhibit growth retardation, short stature, premature graying of hair, hair loss, wrinkling, prematurely aged faces, beaked noses, skin atrophy (wasting away) with scleroderma-like lesions, loss of fat tissues, abnormal fat deposition leading to thin legs and arms, and severe ulcerations around the Achilles tendon and malleoli. Other signs include change in voice, making it weak, hoarse, or high-pitched; atrophy of gonads, leading to reduced fertility; bilateral cataracts (clouding of lens); premature arteriosclerosis (thickening and loss of elasticity of arteries); calcinosis (calcium deposits in blood vessels); atherosclerosis (blockage of blood vessels); type 2 diabetes; loss of bone mass; telangiectasia; and malignancies.[3][9] In fact, the prevalence of rare cancers, such as meningiomas, are increased in individuals with Werner syndrome.[15]
Approximately 90% of individuals with Werner Syndrome have any of a range of mutations in the eponymous gene, WRN, the only gene currently connected to Werner syndrome.[14] WRN encodes the WRNp protein, a 1432 amino acid protein with a central domain resembling members of the RecQ helicases. WRNp is active in unwinding DNA, a step necessary in DNA repair and DNA replication.[10][11] Since WRNp's function depends on DNA, it is only functional when localized to the nucleus.
Mutations that cause Werner syndrome only occur at the regions of the gene that encode for protein and not at non-coding regions.[16] These mutations can have a range of effects. They may decrease the stability of the transcribed messenger RNA (mRNA), which increases the rate at which they are degraded. With fewer mRNA, fewer are available to be translated into the WRNp protein. Mutations may also lead to the truncation (shortening) of the WRNp protein, leading to the loss of its nuclear localization signal sequence, which would normally transport it to the nucleus where it can interact with the DNA. This leads to a reduction in DNA repair.[16] Furthermore, mutated proteins are more likely to be degraded than normal WRNp.[11] Apart from causing defects in DNA repair, its aberrant association with p53 down-regulates the function of p53, leading to a reduction in p53-dependent apoptosis and increase the survival of these dysfunctional cells.[17]
Cells of affected individuals have reduced lifespan in culture,[18] more chromosome breaks and translocations[19] and extensive deletions.[20] These DNA damages, chromosome aberrations and mutations may in turn cause more RecQ-independent aging phenotypes.
Bloom syndrome
Bloom syndrome (BS) is a very rare autosomal recessive disorder.[21] Incidence rates are unknown, although it is known to be higher in people of Ashkenazi Jewish background, presenting in around 1 in 50,000. Approximately one-third of individuals who have BS are of Ashkenazi Jewish descent.
There is no evidence from the Bloom's Syndrome Registry or from the peer-reviewed medical literature that BS is a progeroid condition associated with advanced aging. It is, however, associated with early-onset cancer and adult-type diabetes and also with Werner syndrome, which is a progeroid syndrome, through mutation in the RecQ helicases. These associations have led to the speculation that BS could be associated with aging. Unfortunately, the average lifespan of persons with Bloom syndrome is 27 years; consequently, there is insufficient information to completely rule out the possibility that BS is associated with some features of aging.
People with BS start their life with a low weight and length when they are born. Even as adults, they typically remain under 5 feet tall.[22] Individuals with BS are characterized by low weight and height and abnormal facial features, particularly a long, narrow face with a small lower jaw, a large nose and prominent ears. Most also develop photosensitivity, which causes the blood vessels to be dilated and leads to reddening of the skin, usually presented as a "butterfly-shaped patch of reddened skin across the nose and cheeks".[23] Other characteristics of BS include learning disabilities, an increased risk of diabetes, gastroesophageal reflux (GER), and chronic obstructive pulmonary disease (COPD). GER may also lead to recurrent infections of the upper respiratory tract, ears, and lungs during infancy. BS causes infertility in males and reduced fertility and early-onset menopause in females. In line with any RecQ-associated PS, people with BS have an increased risk of developing cancer, often more than one type.
BS is caused by mutations in the BLM gene, which encodes for the Bloom syndrome protein, a RecQ helicase.[24] These mutations may be frameshift, missense, non-sense, or mutations of other kinds and are likely to cause deletions in the gene product.[25][26] Apart from helicase activity that is common to all RecQ helices, it also acts to prevent inappropriate homologous recombination. During replication of the genome, the two copies of DNA, called sister chromatids, are held together through a structure called the centromere. During this time, the homologous (corresponding) copies are in close physical proximity to each other, allowing them to 'cross' and exchange genetic information, a process called homologous recombination. Defective homologous recombination can cause mutation and genetic instability.[27] Such defective recombination can introduce gaps and breaks within the genome and disrupt the function of genes, possibly causing growth retardation, aging and elevated risk of cancer. It introduces gaps and breaks within the genome and disrupts the function of genes, often causing retardation of growth, aging and elevated risks of cancers. The Bloom syndrome protein interacts with other proteins, such as topoisomerase IIIα and RMI2,[28][29][30] and suppresses illegitimate recombination events between sequences that are divergent from strict homology, thus maintaining genome stability.[27] Individuals with BS have a loss-of-function mutation, which means that the illegitimate recombination is no longer suppressed, leading to higher rates of mutation (~10-100 times above normal, depending on cell type).[31][32]
NER protein-associated PS
Nucleotide excision repair is a DNA repair mechanism. There are three excision repair pathways: nucleotide excision repair (NER), base excision repair (BER), and DNA mismatch repair (MMR). In NER, the damaged DNA strand is removed and the undamaged strand is kept as a template for the formation of a complementary sequence with DNA polymerase. DNA ligase joins the strands together to form dsDNA. There are two subpathways for NER, which differ only in their mechanism for recognition: global genomic NER (GG-NER) and transcription coupled NER (TC-NER).
Defects in the NER pathway have been linked to progeroid syndromes. There are 28 genes in this pathway. Individuals with defects in these genes often have developmental defects and exhibit neurodegeneration. They can also develop CS, XP, and TTD,[33] often in combination with each other, as with combined xeroderma pigmentosa-Cockayne syndrome (XP-CS).[34] Variants of these diseases, such as DeSanctis–Cacchione syndrome and Cerebro-oculo-facio-skeletal (COFS) syndrome, can also be caused by defects in the NER pathway. However, unlike RecQ-associated PS, not all individuals affected by these diseases have increased risk of cancer.[3] All these disorders can be caused by mutations in a single gene, XPD,[35][36][37][38] or in other genes.[39]
Cockayne syndrome
Cockayne syndrome (CS) is a rare autosomal recessive PS. There are three types of CS, distinguished by severity and age of onset. It occurs at a rate of about 1 in 300,000-500,000 in the United States and Europe.[40] [41] The mean age of death is ~12 years,[42] although the different forms differ significantly. Individuals with the type I (or classical) form of the disorder usually first show symptoms between one and three years and have lifespans of between 20 and 40 years. Type II Cockayne syndrome (CSB) is more severe: symptoms present at birth and individuals live to approximately 6–7 years of age.[3] Type III has the mildest symptoms, first presents later in childhood,[41] and the cause of death is often severe nervous system deterioration and respiratory tract infections.[43]
Individuals with CS appear prematurely aged and exhibit severe growth retardation leading to short stature. They have a small head (less than the -3 standard deviation),[44] fail to gain weight and failure to thrive. They also have extreme cutaneous photosensitivity (sensitivity to sunlight), neurodevelopmental abnormalities, and deafness, and often exhibit lipoatrophy, atrophic skin, severe tooth decay, sparse hair, calcium deposits in neurons, cataracts, sensorineural hearing loss, pigmentary retinopathy, and bone abnormalities. However, they do not have a higher risk of cancer.
Type I and II are known to be caused by mutation of a specific gene. CSA is caused by mutations in the cross-complementing gene 8 (ERCC8), which encodes for the CSA protein. These mutations are thought to cause alternate splicing of the pre-mRNA which leads to an abnormal protein.[45] CSB is caused by mutations in the ERCC6 gene, which encodes the CSB protein.[46] CSA and CSB are involved in transcription-coupled NER (TC-NER), which is involved in repairing DNA; they ubiquitinate RNA polymerase II, halting its progress thus allowing the TC-NER mechanism to be carried out.[47] The ubiquitinated RNAP II then dissociates and is degraded via the proteasome.[48] Mutations in ERCC8, ERCC6, or both mean DNA is no longer repaired through TC-NER, and the accumulation of mutations leads to cell death, which may contribute to the symptoms of Cockayne syndrome.[41]
Xeroderma pigmentosum
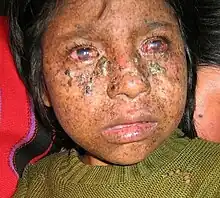
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder, affecting about one per million in the United States and autochthonic Europe populations[40] but with a higher incidence rate in Japan, North Africa, and the Middle East.[50] There have been 830 published cases from 1874 to 1982.[51] The disorder presents at infancy or early childhood.
Xeroderma pigmentosum mostly affects the eye and skin. Individuals with XP have extreme sensitivity to light in the ultraviolet range starting from one to two years of age,[51] and causes sunburn, freckling of skin, dry skin and pigmentation after exposure.[52] When the eye is exposed to sunlight, it becomes irritated and bloodshot, and the cornea becomes cloudy. Around 30% of affected individuals also develop neurological abnormalities, including deafness, poor coordination, decreased intellectual abilities, difficulty swallowing and talking, and seizures; these effects tend to become progressively worse over time. All affected individuals have a 1000-fold higher risk of developing skin cancer:[53] half of the affected population develop skin cancer by age 10, usually at areas most exposed to sunlight (e.g. face, head, or neck).[54] The risk for other cancers such as brain tumors, lung cancer and eye cancers also increase.[55]
There are eight types of XP (XP-A through XP-G), plus a variant type (XP-V), all categorized based on the genetic cause. XP can be caused by mutations in any of these genes: DDB2, ERCC2, ERCC3, ERCC4, ERCC5, XPA, XPC. These genes are all involved in the NER repair pathway that repairs damaged DNA. The variant form, XP-V, is caused by mutations in the POLH gene, which unlike the rest does not code for components of the NER pathway but produces a DNA polymerase that allows accurate translesion synthesis of DNA damage resulting from UV radiation; its mutation leads to an overall increase in UV-dependent mutation, which ultimately causes the symptoms of XP.
Trichothiodystrophy
Trichothiodystrophy (TTD) is a rare autosomal recessive disease whose symptoms span across multiple systems[56] and can vary greatly in severity. The incidence rate of TTD is estimated to be 1.2 per million in Western Europe.[40] Milder cases cause sparse and brittle hair, which is due to the lack of sulfur,[57] an element that is part of the matrix proteins that give hair its strength.[58] More severe cases cause delayed development, significant intellectual disability, and recurrent infection; the most severe cases see death at infancy or early childhood.
TTD also affects the mother of the affected child during pregnancy, when she may experience pregnancy-induced high blood pressure and develop HELLP syndrome. The baby has a high risk of being born prematurely and will have a low birth weight. After birth, the child's normal growth is retarded, resulting in a short stature.
Other symptoms include scaly skin, abnormalities of the fingernails and toenails, clouding of the lens of the eye from birth (congenital cataracts), poor co-ordination, and ocular and skeletal abnormalities. Half of affected individuals also experience photosensitivity to UV light.[56]
TTD is caused by mutations in one of three genes, ERCC2, ERCC3, or GTF2H5, the first two of which are also linked to xeroderma pigmentosum. However, patients with TTD do not show a higher risk of developing skin cancer, in contrast to patients with XP.[57] The three genes associated with TTD encode for XPB, XPD and p8/TTDA of the general transcription factor IIH (TFIIH) complex,[59] which is involved in transcription and DNA damage repair. Mutations in one of these genes cause reduction of gene transcription, which may be involved in development (including placental development),[60] and thus may explain retardation in intellectual abilities, in some cases;[57] these mutations also lead to reduction in DNA repair, causing photosensitivity.[57][61]
A form of TTD without photosensitivity also exists, although its mechanism is unclear. The MPLKIP gene has been associated with this form of TTD, although it accounts for only 20% of all known cases of the non-photosensitive form of TTD, and the function of its gene product is also unclear. Mutations in the TTDN1 gene explain another 10% of non-photosensitive TTD.[62] The function of the gene product of TTDN1 is unknown, but the sex organs of individuals with this form of TTD often produce no hormones, a condition known as hypogonadism.[62]
Defects in Lamin A/C

Hutchinson–Gilford progeria syndrome (HGPS) and restrictive dermopathy (RD) are two PS caused by a defect in lamin A/C, which is encoded by the LMNA gene.[63][64] Lamin A is a major nuclear component that determines the shape and integrity of the nucleus, by acting as a scaffold protein that forms a filamentous meshwork underlying the inner nuclear envelope, the membrane that surrounds the nucleus.
Hutchinson–Gilford progeria syndrome
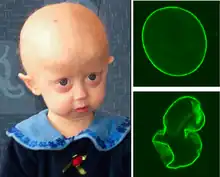
Hutchinson–Gilford progeria syndrome is an extremely rare developmental autosomal dominant condition, characterized by premature and accelerated aging (~7 times the normal rate)[65] beginning at childhood. It affects 1 in ~4 million newborns; over 130 cases have been reported in the literature since the first described case in 1886.[66] The mean age of diagnosis is ~3 years and the mean age of death is ~13 years. The cause of death is usually myocardial infarction, caused by the severe hardening of the arteries (arteriosclerosis).[67] There is currently no treatment available.[68]
Individuals with HGPS typically appear normal at birth, but their growth is severely retarded, resulting in short stature, a very low body weight and delayed tooth eruption. Their facial/cranial proportions and facial features are abnormal, characterized by larger-than-normal eyes, a thin, beaked nose, thin lips, small chin and jaw (micrognathia), protruding ears, scalp hair, eyebrows, and lashes, hair loss, large head, large fontanelle and generally appearing aged. Other features include skeletal alterations (osteolysis, osteoporosis), amyotrophy (wasting of muscle), lipodystrophy and skin atrophy (loss of subcutaneous tissue and fat) with sclerodermatous focal lesions, severe atherosclerosis and prominent scalp veins.[69] However, the level of cognitive function, motor skills, and risk of developing cancer is not affected significantly.[66]
HGPS is caused by sporadic mutations (not inherited from parent) in the LMNA gene, which encodes for lamin A.[63][64] Specifically, most HGPS are caused by a dominant, de novo, point mutation p.G608G (GGC > GGT).[64] This mutation causes a splice site within exon 11 of the pre-mRNA to come into action, leading to the last 150 base pairs of that exon, and consequently, the 50 amino acids near the C-terminus, being deleted.[64] This results in a truncated lamin A precursor (a.k.a. progerin or LaminAΔ50).[70]
After being translated, a farnesol is added to prelamin A using protein farnesyltransferase; this farnesylation is important in targeting lamin to the nuclear envelope, where it maintains its integrity. Normally, lamin A is recognized by ZMPSTE24 (FACE1, a metalloprotease) and cleaved, removing the farnesol and a few other amino acids.
In the truncated lamin A precursor, this cleavage is not possible and the prelamin A cannot mature. When the truncated prelamin A is localized to the nuclear envelope, it will not be processed and accumulates,[71] leading to "lobulation of the nuclear envelope, thickening of the nuclear lamina, loss of peripheral heterochromatin, and clustering of nuclear pores", causing the nucleus to lose its shape and integrity.[72] The prelamin A also maintains the farnesyl and a methyl moiety on its C-terminal cysteine residue, ensuring their continued localization at the membrane. When this farnesylation is prevented using farnesyltransferase inhibitor (FTI), the abnormalities in nuclear shape are significantly reduced.[71][73]
HGPS is considered autosomal dominant, which means that only one of the two copies of the LMNA gene needs to be mutated to produce this phenotype. As the phenotype is caused by an accumulation of the truncated prelamin A, only mutation in one of the two genes is sufficient.[72] At least 16 Other mutations in lamin A/C,[74][75] or defects in the ZMPSTE24 gene,[76] have been shown to cause HGPS and other progeria-like symptoms, although these are less studied.
Repair of DNA double-strand breaks can occur by one of two processes, non-homologous end joining (NHEJ) or homologous recombination (HR). A-type lamins promote genetic stability by maintaining levels of proteins which have key roles in NHEJ and HR.[77] Mouse cells deficient for maturation of prelamin A show increased DNA damage and chromosome aberrations and have increased sensitivity to DNA damaging agents.[78] In HGPS, the inability to adequately repair DNA damages due to defective A-type lamin may cause aspects of premature aging (see DNA damage theory of aging).
Restrictive dermopathy
Restrictive dermopathy (RD), also called tight skin contracture syndrome, is a rare, lethal autosomal recessive perinatal genodermatosis.[79] Two known causes of RD are mutations in the LMNA gene, which lead to the production of truncated prelamin A precursor, and insertions in the ZMPSTE24, which lead to a premature stop codon.[79]
Individuals with RD exhibit growth retardation starting in the uterus, tight and rigid skin with erosions, prominent superficial vasculature and epidermal hyperkeratosis, abnormal facial features (small mouth, small pinched nose and micrognathia), sparse or absent eyelashes and eyebrows, mineralization defects of the skull, thin dysplastic clavicles, pulmonary hypoplasia and multiple joint contractures. Most affected individuals die in the uterus or are stillbirths, and liveborns usually die within a week.
Defects in FBN1
Patients with Marfan-progeroid-lipodystrophy syndrome typically exhibit congenital lipodystrophy and a neonatal progeroid appearance.[80][81] Sometimes identified as having neonatal progeroid syndrome, the term is a misnomer since they do not exhibit accelerated aging.[82] The condition is caused by mutations near the 3'-terminus of the FBN1 gene.[80][81][82][83][84][85]
A common cause for premature aging
Hutchinson–Gilford progeria syndrome, Werner syndrome, and Cockayne syndrome are the three genetic disorders in which patients have premature aging features. Premature aging also develops on some animal models which have genetic alterations.[86][87] Although the patients with these syndromes and the animal models with premature aging symptoms have different genetic backgrounds, they all have abnormal structures of tissues/organs as a result of defective development. Misrepair-accumulation aging theory[88][89] suggests that the abnormality of tissue structure is the common point between premature aging and normal aging.[90] Premature aging is a result of Mis-construction during development as a consequence of gene mutations, whereas normal aging is a result of accumulation of Misrepairs for the survival of an organism. Thus the process of development and that of aging are coupled by Mis-construction and Mis-re-construction (Misrepair) of the structure of an organism.
Unknown causes
Wiedemann–Rautenstrauch syndrome
Wiedemann–Rautenstrauch (WR) syndrome, also known as neonatal progeroid syndrome,[91] is an autosomal recessive progeroid syndrome. More than 30 cases have been reported.[92] Most affected individuals die by seven months of age, but some do survive into their teens.
WR is associated with abnormalities in bone maturation, and lipids and hormone metabolism.[93] Affected individuals exhibit intrauterine and postnatal growth retardation, leading to short stature and an aged appearance from birth. They have physical abnormalities including a large head (macrocephaly), sparse hair, prominent scalp veins, inward-folded eyelids, widened anterior fontanelles, hollow cheeks (malar hypoplasia), general loss of fat tissues under the skin, delayed tooth eruption, abnormal hair pattern, beaked noses, mild to severe intellectual disability and dysmorphism.[94]
The cause of WR is unknown, although defects in DNA repair have been implicated.[92]
Rothmund–Thomson syndrome
Classified as an autosomal recessive defect, but the pathology has still yet to be well researched.
Cancer
Some segmental progeroid syndromes, such as Werner syndrome (WS), Bloom syndrome (BS), Rothmund-Thomson syndromes (RTS) and combined xeroderma pigmentosa-Cockayne syndrome (XP-CS), are associated with an increased risk of developing cancer in the affected individual; two exceptions are Hutchinson–Gilford progeria (HGPS) and Cockayne syndrome.[95]
Animal models
Within animal models for progeroid syndromes, early observations have detected abnormalities within overall mitochondrial function,[96][97] signal transduction between membrane receptors,[98] and nuclear regulatory proteins.
Other
Alterations in lipid and carbohydrate metabolism, a triplet-repeat disorder (myotonic dystrophy) and an idiopathic disorder
Society and popular culture
People
Hayley Okines was an English girl with classic progeria famed for her efforts in spreading awareness of the condition. She was featured in the media.[99]
Lizzie Velásquez is an American motivational speaker who has a syndrome that resembles progeria, although the exact nature is unclear; it is now thought to be a form of neonatal progeroid syndrome.[100] Velásquez is an advocate of anti-bullying.[101][102]
Jesper Sørensen is widely recognized in Denmark as the only child in Denmark and Scandinavia with progeria (as of 2008).[103] His fame came about after a documentary in 2008 on TV 2 about Sørensen.[104]
Literature and Theatre
F. Scott Fitzgerald's 1922 short story The Curious Case of Benjamin Button is about a boy who was born with the appearance of a 70-year-old and who ages backwards. This short story is thought to be inspired by progeria.[105] The description of the fictitious Smallweed family in the Charles Dickens' Bleak House suggests the characters had progeria.[106] Christopher Snow, the main character in Dean Koontz's Moonlight Bay Trilogy, has xeroderma pigmentosum, as does Luke from the 2002 novel Going Out by Scarlett Thomas. In the visual novel Chaos;Head, the character Shogun eventually dies of a progeroid syndrome, and in its sequel Chaos;Child, more characters get this same fictional progeroid syndrome, which by then is called Chaos Child Syndrome. In Kimberly Akimbo, a 2000 play by David Lindsay-Abaire, and its Tony Award for Best Musical-winning adaptation of the same name, the main character, Kimberly Levaco, has an unnamed progeria-like condition.
Film
Paa, a 2009 Indian comedy-drama film, features a protagonist, Auro (Amitabh Bachchan), who has progeria. Jack is a 1996 American comedy-drama film, in which the titular character (portrayed by Robin Williams) has Werner syndrome. Taiyou no Uta, a 2006 Japanese film, features Kaoru Amane (portrayed by Yui), a 16-year-old girl has xeroderma pigmentosum.
See also
- DeSanctis–Cacchione syndrome, an extremely rare variant of xeroderma pigmentosum (XP)
- Dyskeratosis congenita, a rare progressive congenital disorder of the skin and bone marrow in some ways resembling progeria
- Fanconi anemia, a rare genetic defect in a cluster of proteins responsible for DNA repair
- Li–Fraumeni syndrome, a rare autosomal genetic disorder caused by defects in DNA repair
- Nijmegen breakage syndrome, a rare autosomal recessive genetic disorder caused by defect(s) in the Double Holliday junction DNA repair mechanism
- Nestor-Guillermo progeria syndrome, an extremely rare genetic disorder which is unique from other PS because of the absence of any cardiovascular abnormalities (which lead to premature death in cases where they are present)
References
- ↑ Sinha, Jitendra Kumar; Ghosh, Shampa; Raghunath, Manchala (May 2014). "Progeria: a rare genetic premature ageing disorder". Indian J Med Res. 139 (5): 667–74. PMC 4140030. PMID 25027075.
- ↑ Gordon, Leslie B.; Cao, Kan; Collins, Francis S. (2012). "Progeria: Translational insights from cell biology". J Cell Biol. 199 (1): 9–13. doi:10.1083/jcb.201207072. PMC 3461511. PMID 23027899.
- 1 2 Kaneko, H; Fukao, T; Kondo, N (2004). "The function of RecQ helicase gene family (especially BLM) in DNA recombination and joining". Advances in Biophysics. 38 (Complete): 45–64. doi:10.1016/S0065-227X(04)80061-3. PMID 15493327.
- ↑ Mohaghegh, P; Hickson, ID (2001). "DNA helicase deficiencies associated with cancer predisposition and premature ageing disorders". Human Molecular Genetics. 10 (7): 741–6. doi:10.1093/hmg/10.7.741. PMID 11257107.
- ↑ Goode, EL; Ulrich, CM; Potter, JD (2002). "Polymorphisms in DNA repair genes and associations with cancer risk". Cancer Epidemiology, Biomarkers & Prevention. 11 (12): 1513–30. PMID 12496039.
- 1 2 Ouyang, KJ; Woo, LL; Ellis, NA (2008). "Homologous recombination and maintenance of genome integrity: Cancer and aging through the prism of human RecQ helicases". Mechanisms of Ageing and Development. 129 (7–8): 425–40. doi:10.1016/j.mad.2008.03.003. PMID 18430459. S2CID 6804631.
- ↑ Hanada, K.; Hickson, I. D. (2007). "Molecular genetics of RecQ helicase disorders". Cellular and Molecular Life Sciences. 64 (17): 2306–22. doi:10.1007/s00018-007-7121-z. PMID 17571213. S2CID 29287970.
- 1 2 3 Hasty, P.; Campisi, J; Hoeijmakers, J; Van Steeg, H; Vijg, J (2003). "Aging and Genome Maintenance: Lessons from the Mouse?". Science. 299 (5611): 1355–9. doi:10.1126/science.1079161. PMID 12610296. S2CID 840477.
- 1 2 Gray, Matthew D.; Shen, Jiang-Cheng; Kamath-Loeb, Ashwini S.; Blank, A.; Sopher, Bryce L.; Martin, George M.; Oshima, Junko; Loeb, Lawrence A. (1997). "The Werner syndrome protein is a DNA helicase". Nature Genetics. 17 (1): 100–3. doi:10.1038/ng0997-100. PMID 9288107. S2CID 20587915.
- 1 2 3 "Werner syndrome". Genetics Home Reference. Retrieved 18 March 2013.
- ↑ Masala, MV; Scapaticci, S; Olivieri, C; Pirodda, C; Montesu, MA; Cuccuru, MA; Pruneddu, S; Danesino, C; et al. (2007). "Epidemiology and clinical aspects of Werner's syndrome in North Sardinia: Description of a cluster". European Journal of Dermatology. 17 (3): 213–6. doi:10.1684/ejd.2007.0155 (inactive 1 August 2023). PMID 17478382.
{{cite journal}}: CS1 maint: DOI inactive as of August 2023 (link) - 1 2 Epstein, CJ; Martin, GM; Schultz, AL; Motulsky, AG (1966). "Werner's syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process". Medicine. 45 (3): 177–221. doi:10.1097/00005792-196605000-00001. PMID 5327241.
- 1 2 Oshima J, Martin GM, Hisama FM. Werner Syndrome. 2002 Dec 2 [Updated 2012 Dec 13]. In: Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1514/
- ↑ Goto, M; Miller, RW; Ishikawa, Y; Sugano, H (1996). "Excess of rare cancers in Werner syndrome (adult progeria)". Cancer Epidemiology, Biomarkers & Prevention. 5 (4): 239–46. PMID 8722214.
- 1 2 Huang, S; Lee, L; Hanson, NB; Lenaerts, C; Hoehn, H; Poot, M; Rubin, CD; Chen, DF; et al. (2006). "The spectrum of WRN mutations in Werner syndrome patients". Human Mutation. 27 (6): 558–67. doi:10.1002/humu.20337. PMC 1868417. PMID 16673358.
- ↑ Spillare, EA; Robles, AI; Wang, XW; Shen, JC; Yu, CE; Schellenberg, GD; Harris, CC (1999). "P53-mediated apoptosis is attenuated in Werner syndrome cells". Genes & Development. 13 (11): 1355–60. doi:10.1101/gad.13.11.1355. PMC 316776. PMID 10364153.
- ↑ Martin, GM; Sprague, CA; Epstein, CJ (1970). "Replicative life-span of cultivated human cells. Effects of donor's age, tissue, and genotype". Laboratory Investigation. 23 (1): 86–92. PMID 5431223.
- ↑ Salk, D; Au, K; Hoehn, H; Martin, GM (1981). "Cytogenetics of Werner's syndrome cultured skin fibroblasts: Variegated translocation mosaicism". Cytogenetics and Cell Genetics. 30 (2): 92–107. doi:10.1159/000131596. PMID 7273860.
- ↑ Fukuchi, K; Martin, GM; Monnat Jr, RJ (1989). "Mutator phenotype of Werner syndrome is characterized by extensive deletions". Proceedings of the National Academy of Sciences of the United States of America. 86 (15): 5893–7. Bibcode:1989PNAS...86.5893F. doi:10.1073/pnas.86.15.5893. PMC 297737. PMID 2762303.
- ↑ Karow, JK; Constantinou, A; Li, JL; West, SC; Hickson, ID (2000). "The Bloom's syndrome gene product promotes branch migration of holliday junctions". Proceedings of the National Academy of Sciences of the United States of America. 97 (12): 6504–8. Bibcode:2000PNAS...97.6504K. doi:10.1073/pnas.100448097. PMC 18638. PMID 10823897.
- ↑ "Bloom syndrome". Genetics Home Reference. Bethesda, Maryland: U.S. National Library of Medicine, a division of the National Institutes of Health. 27 October 2014. Retrieved 4 November 2014.
- ↑ "Bloom syndrome". Genetics Home Reference. Retrieved 18 March 2013.
- ↑ Cheok, CF; Bachrati, CZ; Chan, KL; Ralf, C; Wu, L; Hickson, ID (2005). "Roles of the Bloom's syndrome helicase in the maintenance of genome stability" (PDF). Biochemical Society Transactions. 33 (Pt 6): 1456–9. doi:10.1042/BST20051456. PMID 16246145.
- ↑ German, J; Sanz, MM; Ciocci, S; Ye, TZ; Ellis, NA (2007). "Syndrome-causing mutations of the BLM gene in persons in the Bloom's Syndrome Registry". Human Mutation. 28 (8): 743–53. doi:10.1002/humu.20501. PMID 17407155. S2CID 44382072.
- ↑ Amor-Guéret, M; Dubois-d'Enghien, C; Laugé, A; Onclercq-Delic, R; Barakat, A; Chadli, E; Bousfiha, AA; Benjelloun, M; et al. (2008). "Three new BLM gene mutations associated with Bloom syndrome". Genetic Testing. 12 (2): 257–61. doi:10.1089/gte.2007.0119. PMID 18471088.
- 1 2 Wang Y, Li S, Smith K, Waldman BC, Waldman AS (2016). "Intrachromosomal recombination between highly diverged DNA sequences is enabled in human cells deficient in Bloom helicase". DNA Repair (Amst.). 41: 73–84. doi:10.1016/j.dnarep.2016.03.005. PMID 27100209.
- ↑ Xu, D; Guo, R; Sobeck, A; Bachrati, CZ; Yang, J; Enomoto, T; Brown, GW; Hoatlin, ME; et al. (2008). "RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability". Genes & Development. 22 (20): 2843–55. doi:10.1101/gad.1708608. PMC 2569887. PMID 18923082.
- ↑ Singh, TR; Ali, AM; Busygina, V; Raynard, S; Fan, Q; Du, CH; Andreassen, PR; Sung, P; et al. (2008). "BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome". Genes & Development. 22 (20): 2856–68. doi:10.1101/gad.1725108. PMC 2569884. PMID 18923083.
- ↑ Liu, Y; West, SC (2008). "More complexity to the Bloom's syndrome complex". Genes & Development. 22 (20): 2737–42. doi:10.1101/gad.1732808. PMC 2751278. PMID 18923071.
- ↑ Langlois, RG; Bigbee, WL; Jensen, RH; German, J (1989). "Evidence for increased in vivo mutation and somatic recombination in Bloom's syndrome". Proceedings of the National Academy of Sciences of the United States of America. 86 (2): 670–4. Bibcode:1989PNAS...86..670L. doi:10.1073/pnas.86.2.670. PMC 286535. PMID 2911598.
- ↑ Kusunoki, Y; Hayashi, T; Hirai, Y; Kushiro, J; Tatsumi, K; Kurihara, T; Zghal, M; Kamoun, MR; et al. (1994). "Increased rate of spontaneous mitotic recombination in T lymphocytes from a Bloom's syndrome patient using a flow-cytometric assay at HLA-A locus". Japanese Journal of Cancer Research. 85 (6): 610–8. doi:10.1111/j.1349-7006.1994.tb02403.x. PMC 5919530. PMID 8063614.
- ↑ Cleaver, JE; Lam, ET; Revet, I (2009). "Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity". Nature Reviews Genetics. 10 (11): 756–68. doi:10.1038/nrg2663. PMID 19809470. S2CID 2211460.
- ↑ Lehmann, AR (2003). "DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy". Biochimie. 85 (11): 1101–11. doi:10.1016/j.biochi.2003.09.010. PMID 14726016.
- ↑ Graham, John M.; Anyane-Yeboa, Kwame; Raams, Anja; Appeldoorn, Esther; Kleijer, Wim J.; Garritsen, Victor H.; Busch, David; Edersheim, Terri G.; et al. (2001). "Cerebro-Oculo-Facio-Skeletal Syndrome with a Nucleotide Excision–Repair Defect and a Mutated XPD Gene, with Prenatal Diagnosis in a Triplet Pregnancy". The American Journal of Human Genetics. 69 (2): 291–300. doi:10.1086/321295. PMC 1235303. PMID 11443545.
- ↑ Cleaver, JE; Thompson, LH; Richardson, AS; States, JC (1999). "A summary of mutations in the UV-sensitive disorders: Xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy". Human Mutation. 14 (1): 9–22. doi:10.1002/(SICI)1098-1004(1999)14:1<9::AID-HUMU2>3.0.CO;2-6. PMID 10447254. S2CID 24148589.
- ↑ Broughton, B. C.; Berneburg, M; Fawcett, H; Taylor, EM; Arlett, CF; Nardo, T; Stefanini, M; Menefee, E; et al. (2001). "Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinical outcomes of mutations in the XPD gene". Human Molecular Genetics. 10 (22): 2539–47. doi:10.1093/hmg/10.22.2539. PMID 11709541.
- ↑ Lehmann, AR (2001). "The xeroderma pigmentosum group D (XPD) gene: One gene, two functions, three diseases". Genes & Development. 15 (1): 15–23. doi:10.1101/gad.859501. PMID 11156600.
- ↑ Andressoo, J.O.; Hoeijmakers, J.H.J. (2005). "Transcription-coupled repair and premature ageing". Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 577 (1–2): 179–94. doi:10.1016/j.mrfmmm.2005.04.004. PMID 16009385.
- 1 2 3 Kleijer, WJ; Laugel, V; Berneburg, M; Nardo, T; Fawcett, H; Gratchev, A; Jaspers, NG; Sarasin, A; et al. (2008). "Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy". DNA Repair. 7 (5): 744–50. doi:10.1016/j.dnarep.2008.01.014. PMID 18329345.
- 1 2 3 "Cockayne syndrome". Genetics Home Reference. NIH. Retrieved 19 March 2013.
- ↑ Nance, MA; Berry, SA (1992). "Cockayne syndrome: Review of 140 cases". American Journal of Medical Genetics. 42 (1): 68–84. doi:10.1002/ajmg.1320420115. PMID 1308368.
- ↑ Andressoo, JO; Hoeijmakers, JH (2005). "Transcription-coupled repair and premature ageing". Mutation Research. 577 (1–2): 179–94. doi:10.1016/j.mrfmmm.2005.04.004. PMID 16009385.
- ↑ Pasquier, L; Laugel, V; Lazaro, L; Dollfus, H; Journel, H; Edery, P; Goldenberg, A; Martin, D; et al. (2006). "Wide clinical variability among 13 new Cockayne syndrome cases confirmed by biochemical assays". Archives of Disease in Childhood. 91 (2): 178–82. doi:10.1136/adc.2005.080473. PMC 2082700. PMID 16428367.
- ↑ Komatsu, A; Suzuki, S; Inagaki, T; Yamashita, K; Hashizume, K (2004). "A kindred with Cockayne syndrome caused by multiple splicing variants of the CSA gene". American Journal of Medical Genetics Part A. 128A (1): 67–71. doi:10.1002/ajmg.a.30087. PMID 15211661. S2CID 39634500.
- ↑ Bregman, DB; Halaban, R; Van Gool, AJ; Henning, KA; Friedberg, EC; Warren, SL (1996). "UV-induced ubiquitination of RNA polymerase II: A novel modification deficient in Cockayne syndrome cells". Proceedings of the National Academy of Sciences of the United States of America. 93 (21): 11586–90. Bibcode:1996PNAS...9311586B. doi:10.1073/pnas.93.21.11586. PMC 38101. PMID 8876179.
- ↑ Lee, K.-B. (2002). "Transcription-coupled and DNA damage-dependent ubiquitination of RNA polymerase II in vitro". Proceedings of the National Academy of Sciences. 99 (7): 4239–4244. Bibcode:2002PNAS...99.4239L. doi:10.1073/pnas.072068399. PMC 123632. PMID 11904382.
- ↑ Yang, LY; Jiang, H; Rangel, KM (2003). "RNA polymerase II stalled on a DNA template during transcription elongation is ubiquitinated and the ubiquitination facilitates displacement of the elongation complex". International Journal of Oncology. 22 (3): 683–9. PMID 12579324.
- ↑ Medical Biochemistry at a Glance. John Wiley & Sons. 28 November 2011. ISBN 978-1118292402. Retrieved 17 June 2011.
Xeroderma pigmentosa is a rare, autosomal recessive disease caused by a defective UV-specific endonuclease. Patients with mutations are unable to repair DNA damage caused by sunlight and have been described as "children of the night."
- ↑ "Xeroderma pigmentosum". Genetics Home Reference. NIH. Retrieved 20 March 2013.
- 1 2 Kraemer, KH; Lee, MM; Scotto, J (1987). "Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases". Archives of Dermatology. 123 (2): 241–50. doi:10.1001/archderm.123.2.241. PMID 3545087.
- ↑ Hengge, UR; Emmert, S (2009). "Clinical Features of Xeroderma Pigmentosum". Molecular Mechanisms of Xeroderma Pigmentosum. Advances in Experimental Medicine and Biology. Vol. 637. pp. 10–8. doi:10.1007/978-0-387-09599-8_2. ISBN 978-0-387-09598-1. PMID 19181106.
- ↑ Kraemer, KH; Patronas, NJ; Schiffmann, R; Brooks, BP; Tamura, D; Digiovanna, JJ (2007). "Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype–phenotype relationship". Neuroscience. 145 (4): 1388–96. doi:10.1016/j.neuroscience.2006.12.020. PMC 2288663. PMID 17276014.
- ↑ Kraemer, KH; Lee, MM; Andrews, AD; Lambert, WC (1994). "The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm". Archives of Dermatology. 130 (8): 1018–21. doi:10.1001/archderm.130.8.1018. PMID 8053698.
- ↑ Cleaver, JE (2005). "Cancer in xeroderma pigmentosum and related disorders of DNA repair". Nature Reviews. Cancer. 5 (7): 564–73. doi:10.1038/nrc1652. PMID 16069818. S2CID 7414610.
- 1 2 Faghri, S; Tamura, D; Kraemer, KH; Digiovanna, JJ (2008). "Trichothiodystrophy: A systematic review of 112 published cases characterises a wide spectrum of clinical manifestations". Journal of Medical Genetics. 45 (10): 609–21. doi:10.1136/jmg.2008.058743. PMC 3459585. PMID 18603627.
- 1 2 3 4 Itin, PH; Sarasin, A; Pittelkow, MR (2001). "Trichothiodystrophy: Update on the sulfur-deficient brittle hair syndromes". Journal of the American Academy of Dermatology. 44 (6): 891–920, quiz 921–4. doi:10.1067/mjd.2001.114294. PMID 11369901. S2CID 26006150.
- ↑ Reis, PJ (1992). "Variations in the strength of wool fibres - A review". Australian Journal of Agricultural Research. 43 (6): 1337. doi:10.1071/AR9921337.
- ↑ Hashimoto, S; Egly, JM (2009). "Trichothiodystrophy view from the molecular basis of DNA repair/transcription factor TFIIH". Human Molecular Genetics. 18 (R2): R224–30. doi:10.1093/hmg/ddp390. PMID 19808800.
- ↑ Moslehi, R; Signore, C; Tamura, D; Mills, JL; Digiovanna, JJ; Tucker, MA; Troendle, J; Ueda, T; et al. (2010). "Adverse effects of trichothiodystrophy DNA repair and transcription gene disorder on human fetal development". Clinical Genetics. 77 (4): 365–73. doi:10.1111/j.1399-0004.2009.01336.x. PMC 3463936. PMID 20002457.
- ↑ Stefanini, M; Botta, E; Lanzafame, M; Orioli, D (2010). "Trichothiodystrophy: From basic mechanisms to clinical implications". DNA Repair. 9 (1): 2–10. doi:10.1016/j.dnarep.2009.10.005. PMID 19931493.
- 1 2 Morice-Picard, F; Cario-André, M; Rezvani, H; Lacombe, D; Sarasin, A; Taïeb, A (2009). "New clinico-genetic classification of trichothiodystrophy". American Journal of Medical Genetics Part A. 149A (9): 2020–30. doi:10.1002/ajmg.a.32902. PMID 19681155. S2CID 25663092.
- 1 2 De Sandre-Giovannoli, A.; Bernard, R; Cau, P; Navarro, C; Amiel, J; Boccaccio, I; Lyonnet, S; Stewart, CL; et al. (2003). "Lamin a Truncation in Hutchinson–Gilford Progeria". Science. 300 (5628): 2055. doi:10.1126/science.1084125. PMID 12702809. S2CID 33927803.
- 1 2 3 4 Eriksson, M; Brown, WT; Gordon, LB; Glynn, MW; Singer, J; Scott, L; Erdos, MR; Robbins, CM; et al. (2003). "Recurrent de novo point mutations in lamin a cause Hutchinson–Gilford progeria syndrome". Nature. 423 (6937): 293–8. Bibcode:2003Natur.423..293E. doi:10.1038/nature01629. hdl:2027.42/62684. PMC 10540076. PMID 12714972. S2CID 4420150.
- ↑ Collins, Francis. "We need better drugs -- now". TED.com. Retrieved 22 March 2013.
- 1 2 "Hutchinson–Gilford progeria syndrome". Genetics Home Reference. Retrieved 16 March 2013.
- ↑ Hennekam, RC (2006). "Hutchinson–Gilford progeria syndrome: Review of the phenotype". American Journal of Medical Genetics Part A. 140 (23): 2603–24. CiteSeerX 10.1.1.333.3746. doi:10.1002/ajmg.a.31346. PMID 16838330. S2CID 15692098.
- ↑ "Progeria". MedlinePlus. Retrieved 16 March 2013.
- ↑ Jansen, T; Romiti, R (2000). "Progeria infantum (Hutchinson–Gilford syndrome) associated with scleroderma-like lesions and acro-osteolysis: A case report and brief review of the literature". Pediatric Dermatology. 17 (4): 282–5. doi:10.1046/j.1525-1470.2000.01775.x. PMID 10990576. S2CID 20739447.
- ↑ De Sandre-Giovannoli, A; Bernard, R; Cau, P; Navarro, C; Amiel, J; Boccaccio, I; Lyonnet, S; Stewart, CL; et al. (2003). "Lamin a truncation in Hutchinson–Gilford progeria". Science. 300 (5628): 2055. doi:10.1126/science.1084125. PMID 12702809. S2CID 33927803.
- 1 2 Young, S. G.; Meta, M.; Yang, S. H.; Fong, L. G. (2006). "Prelamin a Farnesylation and Progeroid Syndromes". Journal of Biological Chemistry. 281 (52): 39741–5. doi:10.1074/jbc.R600033200. PMID 17090536. S2CID 27614400.
- 1 2 Goldman, RD; Shumaker, DK; Erdos, MR; Eriksson, M; Goldman, AE; Gordon, LB; Gruenbaum, Y; Khuon, S; et al. (2004). "Accumulation of mutant lamin a causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome". Proceedings of the National Academy of Sciences of the United States of America. 101 (24): 8963–8. Bibcode:2004PNAS..101.8963G. doi:10.1073/pnas.0402943101. PMC 428455. PMID 15184648.
- ↑ Toth, J. I. (2005). "Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes". Proceedings of the National Academy of Sciences. 102 (36): 12873–12878. Bibcode:2005PNAS..10212873T. doi:10.1073/pnas.0505767102. PMC 1193538. PMID 16129834.
- ↑ Broers, JL; Ramaekers, FC; Bonne, G; Yaou, RB; Hutchison, CJ (2006). "Nuclear lamins: Laminopathies and their role in premature ageing". Physiological Reviews. 86 (3): 967–1008. doi:10.1152/physrev.00047.2005. PMID 16816143. S2CID 5609417.
- ↑ Verstraeten, VL; Broers, JL; Van Steensel, MA; Zinn-Justin, S; Ramaekers, FC; Steijlen, PM; Kamps, M; Kuijpers, HJ; et al. (2006). "Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin a accumulation". Human Molecular Genetics. 15 (16): 2509–22. doi:10.1093/hmg/ddl172. PMID 16825282.
- ↑ Mazereeuw-Hautier, J; Wilson, LC; Mohammed, S; Smallwood, D; Shackleton, S; Atherton, DJ; Harper, JI (2007). "Hutchinson–Gilford progeria syndrome: Clinical findings in three patients carrying the G608G mutation in LMNA and review of the literature". The British Journal of Dermatology. 156 (6): 1308–14. doi:10.1111/j.1365-2133.2007.07897.x. PMID 17459035. S2CID 25944330.
- ↑ Redwood AB, Perkins SM, Vanderwaal RP, Feng Z, Biehl KJ, Gonzalez-Suarez I, Morgado-Palacin L, Shi W, Sage J, Roti-Roti JL, Stewart CL, Zhang J, Gonzalo S (2011). "A dual role for A-type lamins in DNA double-strand break repair". Cell Cycle. 10 (15): 2549–60. doi:10.4161/cc.10.15.16531. PMC 3180193. PMID 21701264.
- ↑ Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadiñanos J, López-Otín C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KS, Tryggvason K, Zhou Z (2005). "Genomic instability in laminopathy-based premature aging". Nat. Med. 11 (7): 780–5. doi:10.1038/nm1266. PMID 15980864. S2CID 11798376.
- 1 2 Navarro, C. L.; De Sandre-Giovannoli, A; Bernard, R; Boccaccio, I; Boyer, A; Geneviève, D; Hadj-Rabia, S; Gaudy-Marqueste, C; et al. (2004). "Lamin a and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy". Human Molecular Genetics. 13 (20): 2493–503. doi:10.1093/hmg/ddh265. PMID 15317753.
- 1 2 Graul-Neumann LM, Kienitz T, Robinson PN, Baasanjav S, Karow B, Gillesen-Kaesbach G, Fahsold R, Schmidt H, Hoffmann K, Passarge E (2010). "Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3-prime terminus of the FBN1-gene". Am. J. Med. Genet. 152A (11): 2749–2755. doi:10.1002/ajmg.a.33690. PMID 20979188. S2CID 26408208.
- 1 2 Takenouchi T, Hida M, Sakamoto Y, Torii C, Kosaki R, Takahashi T, Kosaki K (2013). "Severe congenital lipodystrophy and a progeroid appearance: Mutation in the penultimate exon of FBN1 causing a recognizable phenotype". Am. J. Med. Genet. A. 161A (12): 3057–62. doi:10.1002/ajmg.a.36157. PMID 24039054. S2CID 22797418.
- 1 2 Romere C, Duerrschmid C, Bournat J, Constable P, Jain M, Xia F, Saha PK, Del Solar M, Zhu B, York B, Sarkar P, Rendon DA, Gaber MW, LeMaire SA, Coselli JS, Milewicz DM, Sutton VR, Butte NF, Moore DD, Chopra AR (April 2016). "Asprosin, a Fasting-Induced Glucogenic Protein Hormone". Cell. 165 (3): 566–79. doi:10.1016/j.cell.2016.02.063. PMC 4852710. PMID 27087445.
- ↑ Garg A, Xing C (2014). "De novo heterozygous FBN1 mutations in the extreme C-terminal region cause progeroid fibrillinopathy". Am. J. Med. Genet. A. 164A (5): 1341–5. doi:10.1002/ajmg.a.36449. PMC 7597435. PMID 24665001. S2CID 42280802.
- ↑ Jacquinet A, Verloes A, Callewaert B, Coremans C, Coucke P, De Paepe A, Kornak U, Lebrun F, Lombret J, Pierard GE, Robinson PN, Symoens S, Van Maldergem L, Debray FG (2014). "Neonatal progeroid variant of Marfan syndrome with congenital lipodystrophy results from mutations at the 3' end of FBN1 gene". Eur. J. Med. Genet. 57 (5): 230–234. doi:10.1016/j.ejmg.2014.02.012. PMID 24613577.
- ↑ "OMIM Entry - #616914 - MARFAN LIPODYSTROPHY SYNDROME; MFLS". omim.org. Retrieved 2016-12-06.
- ↑ Hirai, M; Ohbayashi, T; Horiguchi, M (2007). "Fibulin-5/DANCE has an elastogenic organizer activity that is abrogated by proteolytic cleavage in vivo". J Cell Biol. 176 (7): 1061–71. doi:10.1083/jcb.200611026. PMC 2064089. PMID 17371835.
- ↑ Lanske, B; Razzaque, MS (2007). "Premature aging in klotho mutant mice: cause or consequence?". Ageing Res Rev. 6 (1): 73–9. doi:10.1016/j.arr.2007.02.002. PMC 2896497. PMID 17353153.
- ↑ Wang, Jicun; Michelitsch, Thomas; Wunderlin, Arne; Mahadeva, Ravi (2009). "Aging as a consequence of Misrepair –a novel theory of aging". arXiv:0904.0575 [q-bio.TO].
- ↑ Wang-Michelitsch, Jicun; Michelitsch, Thomas (2015). "Aging as a process of accumulation of Misrepairs". arXiv:1503.07163 [q-bio.TO].
- ↑ Wang-Michelitsch, Jicun; Michelitsch, Thomas (2015). "Premature aging as a consequence of Misconstruction of tissues and organs during development". arXiv:1505.03905 [q-bio.TO].
- ↑ "Wiedemann Rautenstrauch Syndrome". NORD Rare Disease Report Abstract. Archived from the original on 27 March 2013. Retrieved 16 March 2013.
- 1 2 "Wiedemann–Rautenstrauch syndrome". Orphanet. Retrieved 16 March 2013.
- ↑ Arboleda, H; Quintero, L; Yunis, E (1997). "Wiedemann–Rautenstrauch neonatal progeroid syndrome: Report of three new patients". Journal of Medical Genetics. 34 (5): 433–7. doi:10.1136/jmg.34.5.433. PMC 1050956. PMID 9152846.
- ↑ Toriello, HV (1990). "Wiedemann–Rautenstrauch syndrome". Journal of Medical Genetics. 27 (4): 256–7. doi:10.1136/jmg.27.4.256. PMC 1017029. PMID 2325106.
- ↑ Puzianowska-Kuznicka, M; Kuznicki, J (2005). "Genetic alterations in accelerated ageing syndromes. Do they play a role in natural ageing?". The International Journal of Biochemistry & Cell Biology. 37 (5): 947–60. doi:10.1016/j.biocel.2004.10.011. PMID 15743670.
- ↑ Karikkineth, Ajoy C.; Scheibye-Knudsen, Morten; Fivenson, Elayne; Croteau, Deborah L.; Bohr, Vilhelm A. (January 2017). "Cockayne syndrome: Clinical features, model systems and pathways". Ageing Research Reviews. 33: 3–17. doi:10.1016/j.arr.2016.08.002. ISSN 1872-9649. PMC 5195851. PMID 27507608.
- ↑ Okur, Mustafa N.; Fang, Evandro F.; Fivenson, Elayne M.; Tiwari, Vinod; Croteau, Deborah L.; Bohr, Vilhelm A. (December 2020). "Cockayne syndrome proteins CSA and CSB maintain mitochondrial homeostasis through NAD+ signaling". Aging Cell. 19 (12): e13268. doi:10.1111/acel.13268. ISSN 1474-9726. PMC 7744955. PMID 33166073.
- ↑ Evangelisti, Camilla; Cenni, Vittoria; Lattanzi, Giovanna (November 2016). "Potential therapeutic effects of the MTOR inhibitors for preventing ageing and progeria-related disorders". British Journal of Clinical Pharmacology. 82 (5): 1229–1244. doi:10.1111/bcp.12928. ISSN 1365-2125. PMC 5061804. PMID 26952863.
- ↑ Brown, Tara. "Race Against Time". 60 Minutes. MSN. Archived from the original on 20 December 2013. Retrieved 21 March 2013.
- ↑ "The girl who must eat every 15 minutes to stay alive". The Telegraph. London. 28 June 2010. Archived from the original on 30 June 2010. Retrieved 22 March 2013.
- ↑ "Lizzie Velasquez's Lifelong Struggle With Bullying Revealed". Entertainment Tonight. February 1, 2013. Archived from the original on June 8, 2014. Retrieved March 22, 2013.
- ↑ Chan, Amanda L. (13 September 2012). "Lizzie Velasquez, Born Without Adipose Tissue: 'Maybe You Should Stop Staring And Start Learning'". Huffington Post.
- ↑ "Drengen i den gamle krop". 2008-11-20. Retrieved 22 March 2013.
- ↑ "Seerne er vilde med Jesper". 2012-05-30. Retrieved 22 March 2013.
- ↑ Maloney, W. J. (2009). "Hutchinson–Gilford Progeria Syndrome: Its Presentation in F. Scott Fitzgerald's Short Story 'The Curious Case of Benjamin Button' and Its Oral Manifestations". Journal of Dental Research. 88 (10): 873–6. doi:10.1177/0022034509348765. PMID 19783794. S2CID 40615631.
- ↑ Singh, V. (2010). "Reflections for August: Description of a Family with Progeria by Charles Dickens". Neurology. 75 (6): 571. doi:10.1212/WNL.0b013e3181ec7f6c. PMID 20697111. S2CID 219232325.
Further reading
- Riedl, T.; Hanaoka, F; Egly, JM (2003). "The comings and goings of nucleotide excision repair factors on damaged DNA". The EMBO Journal. 22 (19): 5293–303. doi:10.1093/emboj/cdg489. PMC 204472. PMID 14517266.
- Park, CJ; Choi, BS (2006). "The protein shuffle. Sequential interactions among components of the human nucleotide excision repair pathway". The FEBS Journal. 273 (8): 1600–8. doi:10.1111/j.1742-4658.2006.05189.x. PMID 16623697. S2CID 19820776.
- Singh, DK; Ahn, B; Bohr, VA (2009). "Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging". Biogerontology. 10 (3): 235–52. doi:10.1007/s10522-008-9205-z. PMC 2713741. PMID 19083132.
- Mallory, Susan B.; Krafchik, Bernice R.; Bender, Matthew M.; Potocki, Lorraine; Metry, Denise W. (2003). "Cockayne Syndrome". Pediatric Dermatology. 20 (6): 538–40. doi:10.1111/j.1525-1470.2003.20619.x. PMID 14651579. S2CID 39698691.
External links
- Hutchinson–Gilford Progeria Syndrome described in GeneReviews™
- NIH Office of Rare Diseases Research (ORDR) - a National Institutes of Health branch which coordinates and supports rare diseases research
- Orphanet, a reference portal for information on rare diseases and orphan drugs