
Phosphinidenes (IUPAC: phosphanylidenes, formerly phosphinediyls) are low-valent phosphorus compounds analogous to carbenes and nitrenes, having the general structure RP.[1][2] The "free" form of these compounds is conventionally described as having a singly-coordinated phosphorus atom containing only 6 electrons in its valence level.[2] Most phosphinidenes are highly reactive and short-lived, thereby complicating empirical studies on their chemical properties.[3][4] In the last few decades, several strategies have been employed to stabilize phosphinidenes (e.g. π-donation, steric protection, transition metal complexation),[2][3] and researchers have developed a number of reagents and systems that can generate and transfer phosphinidenes as reactive intermediates in the synthesis of various organophosphorus compounds.[5][6][7][8]
Electronic structure
Like carbenes, phosphinidenes can exist in either a singlet state or triplet state, with the triplet state typically being more stable.[2][4] The stability of these states and their relative energy difference (the singlet-triplet energy gap) is dependent on the substituents.

The ground state in the parent phosphinidene (PH) is a triplet that is 22 kcal/mol more stable than the lowest singlet state.[2][9] This singlet-triplet energy gap is considerably larger than that of the simplest carbene methylene (9 kcal/mol).[10]
Ab initio calculations from Nguyen et al. found that alkyl- and silyl-substituted phosphinidenes have triplet ground states, possibly in-part due to a negative hyperconjugation effect that stabilizes the triplet more than the singlet.[4] Substituents containing lone pairs (e.g. -NX2, -OX, -PX2 ,-SX) were found to stabilize the singlet state, presumably by π-donation into an empty phosphorus 3p orbital; in most of these cases, the energies of the lowest singlet and triplet states were close to degenerate.[4] A singlet ground state could be induced in amino- and phosphino-phosphinidenes by introducing bulky β-substituents, which are thought to destabilize the triplet state by distorting the pyramidal geometry through increased nuclear repulsion.[4]
Stable monomeric phosphino-phosphinidene
Bertrand and coworkers synthesized a stable singlet phosphino-phosphinidene compound using extremely bulky substituents.[3] Hitherto, there had been no free singlet phosphinidenes that were characterized by spectroscopy.[3] The authors prepared a chlorodiazaphospholidine with bulky (2,6-bis[(4-tert-butylphenyl)methyl]-4-methylphenyl) groups, and then synthesized the corresponding phosphaketene. Subsequent photolytic decarbonylation of the phosphaketene produced the phosphino-phosphinidene product as a yellow-orange solid that is stable at room temperature but decomposes immediately in the presence of air and moisture.[3] 31P NMR spectroscopy shows assigned product peaks at 80.2 and -200.4 ppm, with a J-coupling constant of JPP = 883.7 Hz. The very high P-P coupling constant is indicative of P-P multiple bond character.[3] The air/water sensitivity and high solubility of this compound prevented characterization by X-ray crystallography.[3]

Density functional theory and Natural bond orbital (NBO) calculations were used to gain insight into the structure and bonding of these phosphino-phosphinidenes. DFT calculations at the M06-2X/Def2-SVP level of theory on the phospino-phosphinidene with bulky 2,6-bis[4-tert-butylphenyl)methyl]-4-methylphenyl groups suggest that the tri-coordinated phosphorus atom exists in a planar environment.[3] Calculations at the M06-2X/def2-TZVPP//M06-2X/def2-SVP level of theory were applied to a simplified model compound with diisopropylphenyl (Dipp) groups so as to reduce the computational cost for detailed NBO analysis.[3] Inspection of the outputted wavefunctions shows that the HOMO and HOMO-1 are P-P π-bonding orbitals and the LUMO is a P-P π*-antibonding orbital.[3] Further evidence of multiple bond character between the phosphorus atoms was provided by natural resonance theory and a large Wiberg bond index (P1-P2: 2.34).[3] Natural population analysis assigned a negative partial charge to the terminal phosphorus atom (-0.34 q) and a positive charge to the tri-coordinated phosphorus atom (1.16 q).[3]

Despite the negative charge on the terminal phosphorus atom, subsequent studies have shown that this particular phosphinidene is electrophilic at the phosphinidene center. This phosphino-phosphinidene reacts with a number of nucleophiles (CO, isocyanides, carbenes, phosphines, etc.) to form phosphinidene-nucleophile adducts[3][11] Upon nucleophilic addition, the tri-coordinated phosphorus atom becomes non-planar, and it is postulated that the driving force of the reaction is provided by the instability of the phosphinidene's planar geometry.[11]

Phospha-Wittig fragmentation

In 1989, Fritz et al. synthesized the phospha-Wittig species shown to the right.[12] Phospha-Wittig compounds can be viewed as a phosphinidene stabilized by a phosphine. These compounds have been given the label of "phospha-Wittig" as they have two dominant resonance structures (a neutral form and a zwitterionic form) that are analogous to those of the phosphonium ylides that are used in the Wittig reaction.
Fritz et al. found that this particular phospha-Wittig reagent thermally decomposes at 20 °C to give tBu2PBr, LiBr, and cyclophosphanes.[12] The authors proposed that the singlet phosphino-phosphinidene tBu2PP was formed as an intermediate in this reaction. Further evidence for this was provided by trapping experiments, where the thermal decomposition of the phospha-Wittig reagent in the presence of 3,4,-dimethyl-1,3-butadiene and cyclohexene gave rise to the products shown in the figure below.[12]

Phosphinidene complexes
Terminal transition-metal-complexed phosphinidenes LnM=P-R are phosphorus analogs of transition metal carbene complexes where L is a spectator ligand. The first terminal phosphinidene complex was reported by Marinetti et al., who observed the formation of the transient species [(OC)5M=P-Ph] during the fragmentation of 7-phosphanorbornadiene molybdenum and tungsten complexes inside a mass spectrometer.[13][14] Soon after, they discovered that these 7-phosphanorbornadiene complexes could be used to transfer the phosphinidene complex [(OC)5M=P-R] to various unsaturated substrates.[14][15]
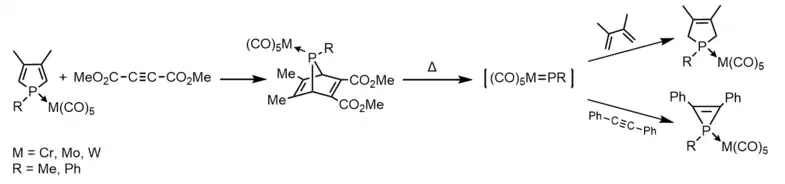
Biskup et al. have reported the synthesis of donor-stabilized terminal electrophilic phosphinidene transition metal complexes via Li/Cl phosphinidenoid complexes[16] which could release free phosphinidene complexes LnM=P-R at mild conditions by P-donor dissociation reactions.[17][18] The phosphinidene complexes decomposed to white phosphorus if no unsaturated substrates were provided.[17]

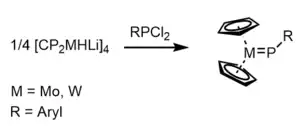
Lappert and coworkers reported the first synthesis of a stable terminal phosphinidene complex: lithium metallocene hydrides [Cp2MHLi]4 of Mo and W were reacted with aryl-dichlorophosphines RPCl2 to yield Cp2M=P-R, which were able to be characterized by single crystal X-ray diffraction.[19]
More common than complexes of terminal phosphinidene ligands are cluster compounds wherein the phosphinidene is a triply and less commonly doubly bridging ligand. One example is the ter-butylphosphinidene complex (t-BuP)Fe3(CO)10.[20]
Dibenzo-7-phosphanorbornadiene derivatives
A class of RPA (A = anthracene) compounds were developed and explored by Cummins and coworkers.[21]
Treatment of a bulky phosphine chloride (RPCl2) with magnesium anthracene affords a dibenzo-7-phosphanorbornadiene compound (RPA).[21] Under thermal conditions, the RPA compound (R = NiPr2) decomposes to yield anthracene; kinetic experiments found this decomposition to be first-order.[21] It was hypothesized that the amino-phosphinidene iPr2NP is formed as a transient intermediate species, and this was corroborated by an experiment where 1,3-cyclohexadiene was used as a trapping agent, forming anti-iPr2NP(C6H8).[21]
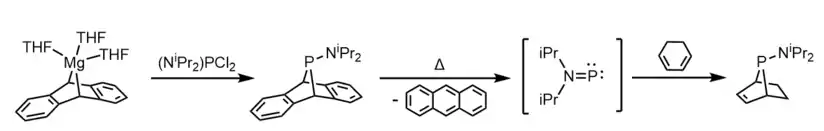
Molecular beam mass spectrometry has enabled the detection of the evolution of amino-phosphinidene fragments from a number of alkylamide derivatives (e.g. Me2NP+ and Me2NPH+ from Me2NPA) in the gas-phase at elevated temperatures.[5]
See also
- Carbene analog
- Phosphorus compounds
References
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "phosphanylidenes". doi:10.1351/goldbook.P04549
- 1 2 3 4 5 Lammertsma, Koop (2003), Majoral, Jean-Pierre (ed.), "Phosphinidenes", New Aspects in Phosphorus Chemistry III, Topics in Current Chemistry, Berlin, Heidelberg: Springer, pp. 95–119, doi:10.1007/b11152, ISBN 978-3-540-36551-8, retrieved 2020-11-02
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Liu, Liu; Ruiz, David A.; Munz, Dominik; Bertrand, Guy (2016). "A Singlet Phosphinidene Stable at Room Temperature". Chem. 1: 147-153. doi:10.1016/j.chempr.2016.04.001.
- 1 2 3 4 5 Nguyen, Minh Tho; Van Keer, Annik; Vanquickenborne, Luc G. (1996). "In Search of Singlet Phosphinidenes". The Journal of Organic Chemistry. 61 (20): 7077–7084. doi:10.1021/jo9604393. ISSN 0022-3263.
- 1 2 Transue, Wesley J.; Velian, Alexandra; Nava, Matthew; García-Iriepa, Cristina; Temprado, Manuel; Cummins, Christopher C. (2017-08-09). "Mechanism and Scope of Phosphinidene Transfer from Dibenzo-7-phosphanorbornadiene Compounds". Journal of the American Chemical Society. 139 (31): 10822–10831. doi:10.1021/jacs.7b05464. hdl:1721.1/117205. ISSN 0002-7863.
- ↑ Hansen, Kerstin; Szilvási, Tibor; Blom, Burgert; Inoue, Shigeyoshi; Epping, Jan; Driess, Matthias (2013-08-14). "A Fragile Zwitterionic Phosphasilene as a Transfer Agent of the Elusive Parent Phosphinidene (:PH)". Journal of the American Chemical Society. 135 (32): 11795–11798. doi:10.1021/ja4072699. ISSN 0002-7863.
- ↑ Krachko, Tetiana; Bispinghoff, Mark; Tondreau, Aaron M.; Stein, Daniel; Baker, Matthew; Ehlers, Andreas W.; Slootweg, J. Chris; Grützmacher, Hansjörg (2017). "Facile Phenylphosphinidene Transfer Reactions from Carbene–Phosphinidene Zinc Complexes". Angewandte Chemie International Edition. 56 (27): 7948–7951. doi:10.1002/anie.201703672. hdl:11245.1/4fe684ed-b624-415c-8873-4a6e9114f66b. ISSN 1521-3773.
- ↑ Pagano, Justin K.; Ackley, Brandon J.; Waterman, Rory (2018-02-21). "Evidence for Iron-Catalyzed α-Phosphinidene Elimination with Phenylphosphine". Chemistry – A European Journal. 24 (11): 2554–2557. doi:10.1002/chem.201704954. ISSN 0947-6539.
- ↑ Benkő, Zoltán; Streubel, Rainer; Nyulászi, László (2006-09-11). "Stability of phosphinidenes—Are they synthetically accessible?". Dalton Transactions (36): 4321–4327. doi:10.1039/B608276A. ISSN 1477-9234.
- ↑ Gronert, Scott; Keeffe, James R.; More O’Ferrall, Rory A. (2011-03-16). "Stabilities of Carbenes: Independent Measures for Singlets and Triplets". Journal of the American Chemical Society. 133 (10): 3381–3389. doi:10.1021/ja1071493. ISSN 0002-7863.
- 1 2 3 Hansmann, Max M.; Jazzar, Rodolphe; Bertrand, Guy (2016-06-30). "Singlet (Phosphino)phosphinidenes are Electrophilic". Journal of the American Chemical Society. 138 (27): 8356–8359. doi:10.1021/jacs.6b04232. ISSN 0002-7863.
- 1 2 3 4 5 Fritz, Gerhard; Vaahs, Tilo; Fleischer, Holm; Matern, Eberhard (1989). "tBu2PPPbrtBu2. LiBr and the Formation of tBu2P". Angewandte Chemie International Edition in English. 28 (3): 315–316. doi:10.1002/anie.198903151. ISSN 1521-3773.
- 1 2 Marinetti, Angela; Mathey, François; Fischer, Jean; Mitschler, André (1982-01-01). "Stabilization of 7-phosphanorbornadienes by complexation; X-ray crystal structure of 2,3-bis(methoxycarbonyl)-5,6-dimethyl-7-phenyl-7-phosphanorbornadiene(pentacarbonyl)-chromium". Journal of the Chemical Society, Chemical Communications (12): 667–668. doi:10.1039/C39820000667. ISSN 0022-4936.
- 1 2 3 Mathey, François (1987). "The Development of a Carbene-like Chemistry with Terminal Phosphinidene Complexes". Angewandte Chemie International Edition in English. 26 (4): 275–286. doi:10.1002/anie.198702753. ISSN 1521-3773.
- 1 2 Marinetti, Angela; Mathey, Francois; Fischer, Jean; Mitschler, Andre (1982-08-01). "Generation and trapping of terminal phosphinidene complexes. Synthesis and x-ray crystal structure of stable phosphirene complexes". Journal of the American Chemical Society. 104 (16): 4484–4485. doi:10.1021/ja00380a029. ISSN 0002-7863.
- ↑ Schmer, Alexander; Junker, Philip; Espinosa Ferao, Arturo; Streubel, Rainer (2021-04-06). "M/X Phosphinidenoid Metal Complex Chemistry". Accounts of Chemical Research. 54 (7): 1754–1765. doi:10.1021/acs.accounts.1c00017. ISSN 0001-4842.
- 1 2 3 Biskup, David; Schnakenburg, Gregor; Boeré, René T.; Espinosa Ferao, Arturo; Streubel, Rainer K. (2023-10-13). "Challenging an old paradigm by demonstrating transition metal-like chemistry at a neutral nonmetal center". Nature Communications. 14 (1): 6456. doi:10.1038/s41467-023-42127-3. ISSN 2041-1723. PMC 10575908. PMID 37833259.
- 1 2 Biskup, David; Schnakenburg, Gregor; Boeré, René T.; Ferao, Arturo Espinosa; Streubel, Rainer (2023-10-03). "A novel access to phosphanylidene–phosphorane complexes via P-donor substitution and a detailed bonding analysis". Dalton Transactions. 52 (38): 13781–13786. doi:10.1039/D3DT02304D. ISSN 1477-9234.
- 1 2 Hitchcock, Peter B.; Lappert, Michael F.; Leung, Wing-Por (1987-01-01). "The first stable transition metal (molybdenum or tungsten) complexes having a metal–phosphorus(III) double bond: the phosphorus analogues of metal aryl- and alkyl-imides; X-ray structure of [Mo(η-C5H5)2(PAr)](Ar = C6H2But3-2,4,6)". Journal of the Chemical Society, Chemical Communications (17): 1282–1283. doi:10.1039/C39870001282. ISSN 0022-4936.
- ↑ Huttner, Gottfried; Knoll, Konrad (1987). "RP-Bridged Metal Carbonyl Clusters: Synthesis, Properties, and Reactions". Angewandte Chemie International Edition in English. 26 (8): 743–760. doi:10.1002/anie.198707431.
- 1 2 3 4 5 Velian, Alexandra; Cummins, Christopher C. (2012-08-20). "Facile Synthesis of Dibenzo-7λ3-phosphanorbornadiene Derivatives Using Magnesium Anthracene". Journal of the American Chemical Society. 134 (34): 13978–13981. doi:10.1021/ja306902j. ISSN 0002-7863.