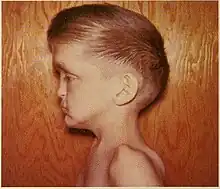
Oto-palato-digital syndrome is the generalised term for two conditions, oto-palato-digital syndrome type I (OPD1) and oto-palato-digital syndrome type II (OPD2), that are both X-linked recessive genetic disorders with overlapping phenotypes. The most severe phenotypes of each syndrome occur only in males, with females generally having attenuated forms of the condition, although this does not apply to all individual cases.[1] Some writers conceptualise oto-palato-digital syndrome as a spectrum disorder including two similarly-presenting genetic syndromes, frontometaphyseal dysplasia and Melnick-Needles syndrome.[2]
The conditions are characterised by skeletal abnormalities, cleft palate (a hole in the roof of the mouth), and hearing loss.[1] These symptoms are common to craniofacial syndromes as a whole.[3] Hand defects are particularly associated.[2] Of the conditions, OPD1 has the milder phenotype, with normal intelligence and modestly reduced stature.[2] In OPD2, the characteristic facial features are more severe and intellectual disability frequent; most OPD2 cases in males are stillborn or die during infancy.[1][2] As an X-linked recessive disorder, both forms are generally more severe in males, who have one X chromosome, than females, who have two.[4] Reports from patients of their experiences demonstrate a broad spectrum of symptom severity, including within families,[5] which has also been reported in the medical literature.[6]
Oto-palato-digital syndrome is caused by a gain-of-function mutation in the FLNA gene on the X chromosome.[1] Women with one copy of the mutation will with each pregnancy have a 50% chance of passing it down to each child.[7] Men with one copy of the mutation will pass it down to all daughters, and will not pass the mutation down to sons.[1] Germline mosaicism for OPD1 has been reported, meaning that unaffected parents with an affected child have a slightly increased risk of bearing another.[2]
The prevalence of oto-palato-digital syndrome is unknown,[7] but estimated to be below 1 in 100,000.[1]
References
- 1 2 3 4 5 6 NORD, Kobylarz Z, Robertson S (2020). "Otopalatodigital Syndrome Type I and II". National Organization for Rare Disorders. Retrieved 3 May 2021.
- 1 2 3 4 5 Robertson SP (23 August 2006). "Otopalatodigital syndrome spectrum disorders: otopalatodigital syndrome types 1 and 2, frontometaphyseal dysplasia and Melnick-Needles syndrome". European Journal of Human Genetics. 15 (1): 3–9. doi:10.1038/sj.ejhg.5201654. PMID 16926860.
- ↑ Sharnetzka R (7 October 2020). "Hearing Loss, Cleft Conditions, and Craniofacial Abnormalities". The Hearing Review. Retrieved 3 May 2021.
- ↑ Call JG, Stern AM, Poznanski AK, Garn SM, Weinstein ED, Hayward JR (January 1972). "Oto-palato-digital syndrome: comparison of clinical and radiographic manifestations in males and females". American Journal of Human Genetics. 24 (1): 24–36. PMC 1762151. PMID 5012690.
- ↑ Arnold J (19 June 2018). "Someone Like Me: Oto-Palatal-Digital Syndrome". Complex Child. Retrieved 3 May 2021.
- ↑ Zaytoun GM, Harboyan G, Kabalan W (February 2002). "The oto-palato-digital syndrome: Variable clinical expressions". Otolaryngology–Head and Neck Surgery. 126 (2): 129–140. doi:10.1067/mhn.2002.122184. PMID 11870342. S2CID 6790951.
- 1 2 Robertson S (3 October 2019). "X-Linked Otopalatodigital Spectrum Disorders". NCBI GeneReviews. University of Washington, Seattle. PMID 20301567. Retrieved 3 May 2021.