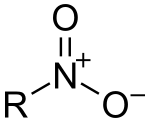
In organic chemistry, nitro compounds are organic compounds that contain one or more nitro functional groups (−NO2). The nitro group is one of the most common explosophores (functional group that makes a compound explosive) used globally. The nitro group is also strongly electron-withdrawing. Because of this property, C−H bonds alpha (adjacent) to the nitro group can be acidic. For similar reasons, the presence of nitro groups in aromatic compounds retards electrophilic aromatic substitution but facilitates nucleophilic aromatic substitution. Nitro groups are rarely found in nature. They are almost invariably produced by nitration reactions starting with nitric acid.[1]
Synthesis
Preparation of aromatic nitro compounds

Aromatic nitro compounds are typically synthesized by nitration. Nitration is achieved using a mixture of nitric acid and sulfuric acid, which produce the nitronium ion (NO+2), which is the electrophile:
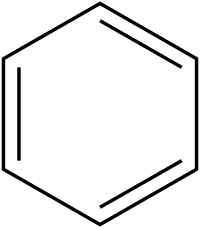 +
+ 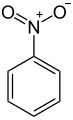
The nitration product produced on the largest scale, by far, is nitrobenzene. Many explosives are produced by nitration including trinitrophenol (picric acid), trinitrotoluene (TNT), and trinitroresorcinol (styphnic acid).[3] Another but more specialized method for making aryl–NO2 group starts from halogenated phenols, is the Zinke nitration.
Preparation of aliphatic nitro compounds
Aliphatic nitro compounds can be synthesized by various methods; notable examples include:
- Free radical nitration of alkanes.[4] The reaction produces fragments from the parent alkane, creating a diverse mixture of products; for instance, nitromethane, nitroethane, 1-nitropropane, and 2-nitropropane are produced by treating propane with nitric acid in the gas phase (e.g. 350–450 °C and 8–12 atm).
- Nucleophilic substitution reactions between halocarbons[5] or organosulfates[6] with silver or alkali nitrite salts.
- Nitromethane can be produced in the laboratory by treating sodium chloroacetate with sodium nitrite.[7]
- Oxidation of oximes[8] or primary amines.[9]
- Reduction of β-nitro alcohols[10] or nitroalkenes.[11]
- By decarboxylation of α-nitro carboxylic acids formed from nitriles and ethyl nitrate.[12][13]
Ter Meer Reaction
In nucleophilic aliphatic substitution, sodium nitrite (NaNO2) replaces an alkyl halide. In the so-called Ter Meer reaction (1876) named after Edmund ter Meer,[14] the reactant is a 1,1-halonitroalkane:
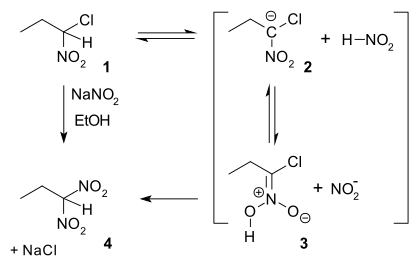
The reaction mechanism is proposed in which in the first slow step a proton is abstracted from nitroalkane 1 to a carbanion 2 followed by protonation to an aci-nitro 3 and finally nucleophilic displacement of chlorine based on an experimentally observed hydrogen kinetic isotope effect of 3.3.[15] When the same reactant is reacted with potassium hydroxide the reaction product is the 1,2-dinitro dimer.[16]
Occurrence
In nature
Chloramphenicol is a rare example of a naturally occurring nitro compound. At least some naturally occurring nitro groups arose by the oxidation of amino groups.[17] 2-Nitrophenol is an aggregation pheromone of ticks.
Examples of nitro compounds are rare in nature. 3-Nitropropionic acid found in fungi and plants (Indigofera). Nitropentadecene is a defense compound found in termites. Nitrophenylethane is found in Aniba canelilla.[18] Nitrophenylethane is also found in members of the Annonaceae, Lauraceae and Papaveraceae.[19]
In pharmaceuticals
Despite the occasional use in pharmaceuticals, the nitro group is associated with mutagenicity and genotoxicity and therefore is often regarded as a liability in the drug discovery process.[20]
Reactions of aliphatic nitro compounds
Reduction
Nitro compounds participate in several organic reactions, the most important being their reduction to the corresponding amines:
- RNO2 + 3 H2 → RNH2 + 2 H2O
Variations on this method is one of the main techniques for arylamine production.
Hydrolysis
In the Nef reaction, a nitro compound hydrolyzes to release a ketone/aldehyde and nitrous oxide.
Acid-base reactions
The α-carbon of nitroalkanes is somewhat acidic. The pKa values of nitromethane and 2-nitropropane are respectively 17.2 and 16.9 in dimethyl sulfoxide (DMSO) solution. These values suggest an aqueous pKa of around 11.[21] In other words, these carbon acids can be deprotonated in aqueous solution. The conjugate base is called a nitronate, they are formed as intermediates in the nitroaldol reaction and Nef reactions.
Condensation reactions
Nitromethane undergoes base-catalyzed additions to aldehydes in 1,2-addition in the nitroaldol reaction. Similarly, it adds to alpha-beta unsaturated carbonyl compounds as a 1,4-addition in the Michael reaction as a Michael donor. Nitroalkenes are Michael acceptors in the Michael reaction with enolate compounds.[22][23]
Biochemical reactions
Many flavin-dependent enzymes are capable of oxidizing aliphatic nitro compounds to less-toxic aldehydes and ketones. Nitroalkane oxidase and 3-nitropropionate oxidase oxidize aliphatic nitro compounds exclusively, whereas other enzymes such as glucose oxidase have other physiological substrates.[24]
Reactions of aromatic nitro compounds
Reduction of aromatic nitro compounds with hydrogen over metal catalysts gives anilines. Virtually all aromatic amines (anilines) are derived from nitroaromatics. A variation is formation of a dimethylaminoarene with palladium on carbon and formaldehyde:[25]
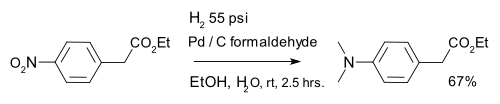
The Leimgruber–Batcho, Bartoli and Baeyer–Emmerling indole syntheses begin with aromatic nitro compounds. Indigo can be synthesized in a condensation reaction from ortho-nitrobenzaldehyde and acetone in strongly basic conditions in a reaction known as the Baeyer–Drewson indigo synthesis.
Explosions
Explosive decomposition of organo nitro compounds are redox reactions, wherein both the oxidant (nitro group) and the fuel (hydrocarbon substituent) are bound within the same molecule. The explosion process generates heat by forming highly stable products including molecular nitrogen (N2), carbon dioxide, and water. The explosive power of this redox reaction is enhanced because these stable products are gases at mild temperatures. Many contact explosives contain the nitro group.
See also
- Functional group
- Reduction of nitro compounds
- Nitration
- Nitrite (also an NO2 group, but bonds differently)
- Nitroalkene
- Nitroglycerin
References
- ↑ Henry Feuer, ed. (1970). Nitro and Nitroso Groups: Part 2, Volume 2. PATAI'S Chemistry of Functional Groups. Vol. 2. John Wiley & Sons Ltd. doi:10.1002/9780470771174. ISBN 978-0-470-77117-4.Saul Patai, ed. (1982). Nitro and Nitroso Groups: Supplement F: Part 2, Volume 2. PATAI'S Chemistry of Functional Groups. John Wiley & Sons Ltd. doi:10.1002/9780470771679. ISBN 978-0-470-77167-9.Saul Patai, ed. (1982). Amino, Nitroso and Nitro Compounds and Their Derivatives: Supplement F: Part 1, Volume 1. PATAI'S Chemistry of Functional Groups. John Wiley & Sons Ltd. doi:10.1002/9780470771662. ISBN 978-0-470-77166-2.
- ↑ Olga V. Dorofeeva; Yuriy V. Vishnevskiy; Natalja Vogt; Jürgen Vogt; Lyudmila V. Khristenko; Sergey V. Krasnoshchekov; Igor F. Shishkov; István Hargittai; Lev V. Vilkov (2007). "Molecular Structure and Conformation of Nitrobenzene Reinvestigated by Combined Analysis of Gas-Phase Electron Diffraction, Rotational Constants, and Theoretical Calculations". Structural Chemistry. 18 (6): 739–753. doi:10.1007/s11224-007-9186-6. S2CID 98746905.
- ↑ Gerald, Booth. "Nitro Compounds, Aromatic". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a17_411. ISBN 978-3527306732.
- ↑ Markofsky, Sheldon; Grace, W.G. (2000). "Nitro Compounds, Aliphatic". Ullmann's Encyclopedia of Industrial Chemistry. doi:10.1002/14356007.a17_401. ISBN 978-3-527-30673-2.
- ↑ Kornblum, N.; Ungnade, H. E. (1963). "1-Nitroöctane". Organic Syntheses. 4: 724. doi:10.15227/orgsyn.038.0075.
- ↑ Walden, P. (1907). "Zur Darstellung aliphatischer Sulfocyanide, Cyanide und Nitrokörper". Berichte der Deutschen Chemischen Gesellschaft. 40 (3): 3214–3217. doi:10.1002/cber.19070400383.
- ↑ Whitmore, F. C.; Whitmore, Marion G. (1923). "Nitromethane". Organic Syntheses. 1: 401. doi:10.15227/orgsyn.003.0083.
- ↑ Olah, George A.; Ramaiah, Pichika; Chang-Soo, Lee; Prakash, Surya (1992). "Convenient Oxidation of Oximes to Nitro Compounds with Sodium Perborate in Glacial Acetic Acid". Synlett. 1992 (4): 337–339. doi:10.1055/s-1992-22006.
- ↑ Ehud, Keinan; Yehuda, Mazur (1977). "Dry ozonation of amines. Conversion of primary amines to nitro compounds". The Journal of Organic Chemistry. 42 (5): 844–847. doi:10.1021/jo00425a017.
- ↑ Chandrasekhar, S.; Shrinidhi, A. (2014). "Useful Extensions of the Henry Reaction: Expeditious Routes to Nitroalkanes and Nitroalkenes in Aqueous Media". Synthetic Communications. 44 (20): 3008–3018. doi:10.1080/00397911.2014.926373. S2CID 98439096.
- ↑ Shrinidhi, A. (2015). "Microwave-assisted chemoselective reduction of conjugated nitroalkenes to nitroalkanes with aqueous tri-n-butyltin hydride". Cogent Chemistry. 1 (1): 1061412. doi:10.1080/23312009.2015.1061412.
- ↑ Wislicenus, Wilhelm; Endres, Anton (1902). "Ueber Nitrirung mittels Aethylnitrat [Nitrification by means of ethyl nitrate]". Berichte der Deutschen Chemischen Gesellschaft. 35 (2): 1755–1762. doi:10.1002/cber.190203502106.
- ↑ Weygand, Conrad (1972). Hilgetag, G.; Martini, A. (eds.). Weygand/Hilgetag Preparative Organic Chemistry (4th ed.). New York: John Wiley & Sons, Inc. p. 1007. ISBN 978-0-471-93749-4.
- ↑ Edmund ter Meer (1876). "Ueber Dinitroverbindungen der Fettreihe". Justus Liebigs Annalen der Chemie. 181 (1): 1–22. doi:10.1002/jlac.18761810102.
- ↑ Hawthorne, M. Frederick (1956). "Aci-Nitroalkanes. I. The Mechanism of the ter Meer Reaction1". Journal of the American Chemical Society. 78 (19): 4980–4984. doi:10.1021/ja01600a048.
- ↑ 3-Hexene, 3,4-dinitro- D. E. Bisgrove, J. F. Brown, Jr., and L. B. Clapp. Organic Syntheses, Coll. Vol. 4, p. 372 (1963); Vol. 37, p. 23 (1957). (Article)
- ↑ Zocher, Georg; Winkler, Robert; Hertweck, Christian; Schulz, Georg E (2007). "Structure and Action of the N-oxygenase AurF from Streptomyces thioluteus". Journal of Molecular Biology. 373 (1): 65–74. doi:10.1016/j.jmb.2007.06.014. PMID 17765264.
- ↑ Maia, José Guilherme S.; Andrade, Eloísa Helena A. (2009). "Database of the Amazon aromatic plants and their essential oils" (PDF). Química Nova. FapUNIFESP (SciELO). 32 (3): 595–622. doi:10.1590/s0100-40422009000300006. ISSN 0100-4042.
- ↑ Kramer, K.U.; Kubitzki, K.; Rohwer, J.G.; Bittrich, V. (1993). Flowering Plants, Dicotyledons: Magnoliid, Hamamelid, and Caryophyllid Families. Families and genera of vascular plants. Springer-Verlag, Berlin. ISBN 978-3-540-55509-4.
- ↑ Nepali K, Lee HY, Liou JP (March 2019). "Nitro-Group-Containing Drugs". J. Med. Chem. 62 (6): 2851–2893. doi:10.1021/acs.jmedchem.8b00147. PMID 30295477. S2CID 52931949.
- ↑ Bordwell, Frederick G; Satish, A. V (1994). "Is Resonance Important in Determining the Acidities of Weak Acids or the Homolytic Bond Dissociation Enthalpies (BDEs) of Their Acidic H-A Bonds?". Journal of the American Chemical Society. 116 (20): 8885. doi:10.1021/ja00099a004.
- ↑ Ranganathan, Darshan; Rao, Bhushan; Ranganathan, Subramania; Mehrotra, Ashok & Iyengar, Radha (1980). "Nitroethylene: a stable, clean, and reactive agent for organic synthesis". The Journal of Organic Chemistry. 45 (7): 1185–1189. doi:10.1021/jo01295a003.
- ↑ Jubert, Carole & Knochel, Paul (1992). "Preparation of polyfunctional nitro olefins and nitroalkanes using the copper-zinc reagents RCu(CN)ZnI". The Journal of Organic Chemistry. 57 (20): 5431–5438. doi:10.1021/jo00046a027.
- ↑ Nagpal, Akanksha; Valley, Michael P.; Fitzpatrick, Paul F.; Orville, Allen M. (2006). "Crystal Structures of Nitroalkane Oxidase: Insights into the Reaction Mechanism from a Covalent Complex of the Flavoenzyme Trapped during Turnover". Biochemistry. 45 (4): 1138–50. doi:10.1021/bi051966w. PMC 1855086. PMID 16430210.
- ↑ "ETHYL p-DIMETHYLAMINOPHENYLACETATE" (PDF). Organic Syntheses. 47: 69. 1967. doi:10.15227/orgsyn.047.0069.