Neuropharmacology is the study of how drugs affect function in the nervous system, and the neural mechanisms through which they influence behavior.[1] There are two main branches of neuropharmacology: behavioral and molecular. Behavioral neuropharmacology focuses on the study of how drugs affect human behavior (neuropsychopharmacology), including the study of how drug dependence and addiction affect the human brain.[2] Molecular neuropharmacology involves the study of neurons and their neurochemical interactions, with the overall goal of developing drugs that have beneficial effects on neurological function. Both of these fields are closely connected, since both are concerned with the interactions of neurotransmitters, neuropeptides, neurohormones, neuromodulators, enzymes, second messengers, co-transporters, ion channels, and receptor proteins in the central and peripheral nervous systems. Studying these interactions, researchers are developing drugs to treat many different neurological disorders, including pain, neurodegenerative diseases such as Parkinson's disease and Alzheimer's disease, psychological disorders, addiction, and many others.
History
Neuropharmacology did not appear in the scientific field until, in the early part of the 20th century, scientists were able to figure out a basic understanding of the nervous system and how nerves communicate between one another. Before this discovery, there were drugs that had been found that demonstrated some type of influence on the nervous system. In the 1930s, French scientists began working with a compound called phenothiazine in the hope of synthesizing a drug that would be able to combat malaria. Though this drug showed very little hope in the use against malaria-infected individuals, it was found to have sedative effects along with what appeared to be beneficial effects toward patients with Parkinson's disease. This black box method, wherein an investigator would administer a drug and examine the response without knowing how to relate drug action to patient response, was the main approach to this field, until, in the late 1940s and early 1950s, scientists were able to identify specific neurotransmitters, such as norepinephrine (involved in the constriction of blood vessels and the increase in heart rate and blood pressure), dopamine (the chemical whose shortage is involved in Parkinson's disease), and serotonin (soon to be recognized as deeply connected to depression). In the 1950s, scientists also became better able to measure levels of specific neurochemicals in the body and thus correlate these levels with behavior.[3] The invention of the voltage clamp in 1949 allowed for the study of ion channels and the nerve action potential. These two major historical events in neuropharmacology allowed scientists not only to study how information is transferred from one neuron to another but also to study how a neuron processes this information within itself.
Overview
Neuropharmacology is a very broad region of science that encompasses many aspects of the nervous system from single neuron manipulation to entire areas of the brain, spinal cord, and peripheral nerves. To better understand the basis behind drug development, one must first understand how neurons communicate with one another.
Neurochemical interactions

To understand the potential advances in medicine that neuropharmacology can bring, it is important to understand how human behavior and thought processes are transferred from neuron to neuron and how medications can alter the chemical foundations of these processes.
Neurons are known as excitable cells because on its surface membrane there are an abundance of proteins known as ion-channels that allow small charged particles to pass in and out of the cell. The structure of the neuron allows chemical information to be received by its dendrites, propagated through the perikaryon (cell body) and down its axon, and eventually passing on to other neurons through its axon terminal. These voltage-gated ion channels allow for rapid depolarization throughout the cell. This depolarization, if it reaches a certain threshold, will cause an action potential. Once the action potential reaches the axon terminal, it will cause an influx of calcium ions into the cell. The calcium ions will then cause vesicles, small packets filled with neurotransmitters, to bind to the cell membrane and release its contents into the synapse. This cell is known as the pre-synaptic neuron, and the cell that interacts with the neurotransmitters released is known as the post-synaptic neuron. Once the neurotransmitter is released into the synapse, it can either bind to receptors on the post-synaptic cell, the pre-synaptic cell can re-uptake it and save it for later transmission, or it can be broken down by enzymes in the synapse specific to that certain neurotransmitter. These three different actions are major areas where drug action can affect communication between neurons.[3]
There are two types of receptors that neurotransmitters interact with on a post-synaptic neuron. The first types of receptors are ligand-gated ion channels or LGICs. LGIC receptors are the fastest types of transduction from chemical signal to electrical signal. Once the neurotransmitter binds to the receptor, it will cause a conformational change that will allow ions to directly flow into the cell. The second types are known as G-protein-coupled receptors or GPCRs. These are much slower than LGICs due to an increase in the amount of biochemical reactions that must take place intracellularly. Once the neurotransmitter binds to the GPCR protein, it causes a cascade of intracellular interactions that can lead to many different types of changes in cellular biochemistry, physiology, and gene expression. Neurotransmitter/receptor interactions in the field of neuropharmacology are extremely important because many drugs that are developed today have to do with disrupting this binding process.[4]
Molecular neuropharmacology
Molecular neuropharmacology involves the study of neurons and their neurochemical interactions, and receptors on neurons, with the goal of developing new drugs that will treat neurological disorders such as pain, neurodegenerative diseases, and psychological disorders (also known in this case as neuropsychopharmacology). There are a few technical words that must be defined when relating neurotransmission to receptor action:
- Agonist – a molecule that binds to a receptor protein and activates that receptor
- Competitive antagonist – a molecule that binds to the same site on the receptor protein as the agonist, preventing activation of the receptor
- Non-competitive antagonist – a molecule that binds to a receptor protein on a different site than that of the agonist, but causes a conformational change in the protein that does not allow activation.
The following neurotransmitter/receptor interactions can be affected by synthetic compounds that act as one of the three above. Sodium/potassium ion channels can also be manipulated throughout a neuron to induce inhibitory effects of action potentials.
GABA
The GABA neurotransmitter mediates the fast synaptic inhibition in the central nervous system. When GABA is released from its pre-synaptic cell, it will bind to a receptor (most likely the GABAA receptor) that causes the post-synaptic cell to hyperpolarize (stay below its action potential threshold). This will counteract the effect of any excitatory manipulation from other neurotransmitter/receptor interactions.
This GABAA receptor contains many binding sites that allow conformational changes and are the primary target for drug development. The most common of these binding sites, benzodiazepine, allows for both agonist and antagonist effects on the receptor. A common drug, diazepam, acts as an allosteric enhancer at this binding site.[5] Another receptor for GABA, known as GABAB, can be enhanced by a molecule called baclofen. This molecule acts as an agonist, therefore activating the receptor, and is known to help control and decrease spastic movement.
Dopamine
The dopamine neurotransmitter mediates synaptic transmission by binding to five specific GPCRs. These five receptor proteins are separated into two classes due to whether the response elicits an excitatory or inhibitory response on the post-synaptic cell. There are many types of drugs, legal and illegal, that affect dopamine and its interactions in the brain. With Parkinson's disease, a disease that decreases the amount of dopamine in the brain, the dopamine precursor Levodopa is given to the patient due to the fact that dopamine cannot cross the blood–brain barrier and L-dopa can. Some dopamine agonists are also given to Parkinson's patients that have a disorder known as restless leg syndrome or RLS. Some examples of these are ropinirole and pramipexole.[6]
Psychological disorders like that of attention deficit hyperactivity disorder (ADHD) can be treated with drugs like methylphenidate (also known as Ritalin), which block the re-uptake of dopamine by the pre-synaptic cell, thereby providing an increase of dopamine left in the synaptic gap. This increase in synaptic dopamine will increase binding to receptors of the post-synaptic cell. This same mechanism is also used by other illegal and more potent stimulant drugs such as cocaine.
Serotonin
The neurotransmitter serotonin has the ability to mediate synaptic transmission through either GPCR's or LGIC receptors. The excitatory or inhibitory post-synaptic effects of serotonin are determined by the type of receptor expressed in a given brain region. The most popular and widely used drugs for the regulation of serotonin during depression are known as SSRIs or selective serotonin reuptake inhibitors. These drugs inhibit the transport of serotonin back into the pre-synaptic neuron, leaving more serotonin in the synaptic gap.
Before the discovery of SSRIs, there were also drugs that inhibited the enzyme that breaks down serotonin. MAOIs or monoamine oxidase inhibitors increased the amount of serotonin in the synapse, but had many side-effects including intense migraines and high blood pressure. This was eventually linked to the drugs interacting with a common chemical known as tyramine found in many types of food.[7]
Ion channels
Ion channels located on the surface membrane of the neuron allows for an influx of sodium ions and outward movement of potassium ions during an action potential. Selectively blocking these ion channels will decrease the likelihood of an action potential to occur. The drug riluzole is a neuroprotective drug that blocks sodium ion channels. Since these channels cannot activate, there is no action potential, and the neuron does not perform any transduction of chemical signals into electrical signals and the signal does not move on. This drug is used as an anesthetic as well as a sedative.[8]
Behavioral neuropharmacology
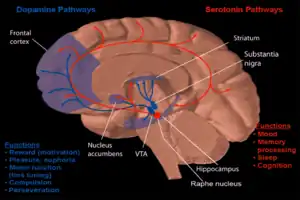
One form of behavioral neuropharmacology focuses on the study of drug dependence and how drug addiction affects the human mind. Most research has shown that the major part of the brain that reinforces addiction through neurochemical reward is the nucleus accumbens. The image to the right shows how dopamine is projected into this area. Long-term excessive alcohol use can cause dependence and addiction. How this addiction occurs is described below.
Ethanol
Alcohol's rewarding and reinforcing (i.e., addictive) properties are mediated through its effects on dopamine neurons in the mesolimbic reward pathway, which connects the ventral tegmental area to the nucleus accumbens (NAcc).[9][10] One of alcohol's primary effects is the allosteric inhibition of NMDA receptors and facilitation of GABAA receptors (e.g., enhanced GABAA receptor-mediated chloride flux through allosteric regulation of the receptor).[11] At high doses, ethanol inhibits most ligand gated ion channels and voltage gated ion channels in neurons as well.[11] Alcohol inhibits sodium-potassium pumps in the cerebellum and this is likely how it impairs cerebellar computation and body co-ordination.[12][13]
With acute alcohol consumption, dopamine is released in the synapses of the mesolimbic pathway, in turn heightening activation of postsynaptic D1 receptors.[9][10] The activation of these receptors triggers postsynaptic internal signaling events through protein kinase A which ultimately phosphorylate cAMP response element binding protein (CREB), inducing CREB-mediated changes in gene expression.[9][10]
With chronic alcohol intake, consumption of ethanol similarly induces CREB phosphorylation through the D1 receptor pathway, but it also alters NMDA receptor function through phosphorylation mechanisms;[9][10] an adaptive downregulation of the D1 receptor pathway and CREB function occurs as well.[9][10] Chronic consumption is also associated with an effect on CREB phosphorylation and function via postsynaptic NMDA receptor signaling cascades through a MAPK/ERK pathway and CAMK-mediated pathway.[10] These modifications to CREB function in the mesolimbic pathway induce expression (i.e., increase gene expression) of ΔFosB in the NAcc,[10] where ΔFosB is the "master control protein" that, when overexpressed in the NAcc, is necessary and sufficient for the development and maintenance of an addictive state (i.e., its overexpression in the nucleus accumbens produces and then directly modulates compulsive alcohol consumption).[10][14][15][16]
Research
Parkinson's disease
Parkinson's disease is a neurodegenerative disease described by the selective loss of dopaminergic neurons located in the substantia nigra. Today, the most commonly used drug to combat this disease is levodopa or L-DOPA. This precursor to dopamine can penetrate through the blood–brain barrier, whereas the neurotransmitter dopamine cannot. There has been extensive research to determine whether L-dopa is a better treatment for Parkinson's disease rather than other dopamine agonists. Some believe that the long-term use of L-dopa will compromise neuroprotection and, thus, eventually lead to dopaminergic cell death. Though there has been no proof, in-vivo or in-vitro, some still believe that the long-term use of dopamine agonists is better for the patient.[17]
Alzheimer's disease
While there are a variety of hypotheses that have been proposed for the cause of Alzheimer's disease, the knowledge of this disease is far from complete to explain, making it difficult to develop methods for treatment. In the brain of Alzheimer's patients, both neuronal nicotinic acetylcholine (nACh) receptors and NMDA receptors are known to be down-regulated. Thus, four anticholinesterases have been developed and approved by the U.S. Food and Drug Administration (FDA) for the treatment in the U.S.A. However, these are not ideal drugs, considering their side-effects and limited effectiveness. One promising drug, nefiracetam, is being developed for the treatment of Alzheimer's and other patients with dementia, and has unique actions in potentiating the activity of both nACh receptors and NMDA receptors.[18]
Future
With advances in technology and our understanding of the nervous system, the development of drugs will continue with increasing drug sensitivity and specificity. Structure–activity relationships are a major area of research within neuropharmacology; an attempt to modify the effect or the potency (i.e., activity) of bioactive chemical compounds by modifying their chemical structures.[8]
See also
References
- ↑ Yeung AWK, Tzvetkov NT, Atanasov AG. When Neuroscience Meets Pharmacology: A Neuropharmacology Literature Analysis. Front Neurosci. 2018 Nov 16;12:852. doi: 10.3389/fnins.2018.00852.
- ↑ Everitt, B. J.; Robbins, T. W. (2005). "Neural systems of reinforcement for drug addiction: from actions to habits to compulsion". Nature Neuroscience. 8 (11): 1481–1489. doi:10.1038/nn1579. PMID 16251991.
- 1 2 Wrobel, S. (2007). "Science, serotonin, and sadness: the biology of antidepressants: A series for the public". The FASEB Journal. 21 (13): 3404–17. doi:10.1096/fj.07-1102ufm. PMID 17967927.
- ↑ Lovinger, D. M. (2008). "Communication Networks in the Brain Neurons, Receptors, Neurotransmitters, and Alcohol. [Review]". Alcohol Research & Health. 31 (3): 196–214.
- ↑ Sigel, E (2002). "Mapping of the benzodiazepine recognition site on GABA(A) receptors". Current Topics in Medicinal Chemistry. 2 (8): 833–9. doi:10.2174/1568026023393444. PMID 12171574.
- ↑ Winkelman, JW; Allen, RP; Tenzer, P; Hening, W (2007). "Restless legs syndrome: nonpharmacologic and pharmacologic treatments". Geriatrics. 62 (10): 13–6. PMID 17922563.
- ↑ López-Muñoz, F.; Alamo, C. (2009). "Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today". Current Pharmaceutical Design. 15 (14): 1563–1586. doi:10.2174/138161209788168001. PMID 19442174.
- 1 2 Narahashi, T (2000). "Neuroreceptors and ion channels as the basis for drug action: past, present, and future". The Journal of Pharmacology and Experimental Therapeutics. 294 (1): 1–26. PMID 10871290.
- 1 2 3 4 5 "Alcoholism – Homo sapiens (human) Database entry". KEGG Pathway. 29 October 2014. Retrieved 9 February 2015.
As one of the primary mediators of the rewarding effects of alcohol, dopaminergic ventral tegmental area (VTA) projections to the nucleus accumbens (NAc) have been identified. Acute exposure to alcohol stimulates dopamine release into the NAc, which activates D1 receptors, stimulating PKA signaling and subsequent CREB-mediated gene expression, whereas chronic alcohol exposure leads to an adaptive downregulation of this pathway, in particular of CREB function. The decreased CREB function in the NAc may promote the intake of drugs of abuse to achieve an increase in reward and thus may be involved in the regulation of positive affective states of addiction. PKA signaling also affects NMDA receptor activity and may play an important role in neuroadaptation in response to chronic alcohol exposure.
- 1 2 3 4 5 6 7 8 Kanehisa Laboratories (29 October 2014). "Alcoholism – Homo sapiens (human)". KEGG Pathway. Retrieved 31 October 2014.
- 1 2 Malenka RC, Nestler EJ, Hyman SE (2009). "Chapter 15: Reinforcement and Addictive Disorders". In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York: McGraw-Hill Medical. p. 372. ISBN 9780071481274.
Despite the high concentrations required for its psychoactive effects, ethanol exerts specific actions on the brain. The initial effects of ethanol result primarily from facilitation of GABAA receptors and inhibition of NMDA glutamate receptors. At higher doses, ethanol also inhibits the functioning of most ligand- and voltage-gated ion channels. It is not known whether ethanol selectively affects these channels via direct low affinity binding or via nonspecific disruption of plasma membranes which then selectively influences these highly complex, multimeric, transmembrane proteins. Ethanol allosterically regulates the GABAA receptor to enhance GABA-activated Cl− flux. The anxiolytic and sedative effects of ethanol, as well as those of barbiturates and benzodiazepines, result from enhancement of GABAergic function. Facilitation of GABAA receptor function is also believed to contribute to the reinforcing effects of these drugs. Not all GABAA receptors are ethanol sensitive. ... Ethanol also acts as an NMDA antagonist by allosterically inhibiting the passage of glutamate-activated Na+ and Ca2+ currents through the NMDA receptor. ... The reinforcing effects of ethanol are partly explained by its ability to activate mesolimbic dopamine circuitry, although it is not known whether this effect is mediated at the level of the VTA or NAc. It also is not known whether this activation of dopamine systems is caused primarily by facilitation of GABAA receptors or inhibition of NMDA receptors, or both. Ethanol reinforcement also is mediated in part by ethanol-induced release of endogenous opioid peptides within the mesolimbic dopamine system, although whether the VTA or NAc is the predominant site of such action is not yet known. Accordingly, the opioid receptor antagonist naltrexone reduces ethanol self-administration in animals and is used with modest effect to treat alcoholism in humans.
- ↑ Forrest MD (April 2015). "Simulation of alcohol action upon a detailed Purkinje neuron model and a simpler surrogate model that runs >400 times faster". BMC Neuroscience. 16 (27): 27. doi:10.1186/s12868-015-0162-6. PMC 4417229. PMID 25928094.
- ↑ Forrest, Michael (April 2015). "The neuroscience reason we fall over when drunk". Science 2.0. Retrieved January 2, 2019.
- ↑ Ruffle JK (November 2014). "Molecular neurobiology of addiction: what's all the (Δ)FosB about?". Am J Drug Alcohol Abuse. 40 (6): 428–437. doi:10.3109/00952990.2014.933840. PMID 25083822.
ΔFosB as a therapeutic biomarker
The strong correlation between chronic drug exposure and ΔFosB provides novel opportunities for targeted therapies in addiction (118), and suggests methods to analyze their efficacy (119). Over the past two decades, research has progressed from identifying ΔFosB induction to investigating its subsequent action (38). It is likely that ΔFosB research will now progress into a new era – the use of ΔFosB as a biomarker. If ΔFosB detection is indicative of chronic drug exposure (and is at least partly responsible for dependence of the substance), then its monitoring for therapeutic efficacy in interventional studies is a suitable biomarker (Figure 2). Examples of therapeutic avenues are discussed herein. ...
Conclusions
ΔFosB is an essential transcription factor implicated in the molecular and behavioral pathways of addiction following repeated drug exposure. The formation of ΔFosB in multiple brain regions, and the molecular pathway leading to the formation of AP-1 complexes is well understood. The establishment of a functional purpose for ΔFosB has allowed further determination as to some of the key aspects of its molecular cascades, involving effectors such as GluR2 (87,88), Cdk5 (93) and NFkB (100). Moreover, many of these molecular changes identified are now directly linked to the structural, physiological and behavioral changes observed following chronic drug exposure (60,95,97,102). New frontiers of research investigating the molecular roles of ΔFosB have been opened by epigenetic studies, and recent advances have illustrated the role of ΔFosB acting on DNA and histones, truly as a molecular switch (34). As a consequence of our improved understanding of ΔFosB in addiction, it is possible to evaluate the addictive potential of current medications (119), as well as use it as a biomarker for assessing the efficacy of therapeutic interventions (121,122,124). Some of these proposed interventions have limitations (125) or are in their infancy (75). However, it is hoped that some of these preliminary findings may lead to innovative treatments, which are much needed in addiction. - ↑ Nestler EJ (December 2013). "Cellular basis of memory for addiction". Dialogues Clin Neurosci. 15 (4): 431–443. PMC 3898681. PMID 24459410.
DESPITE THE IMPORTANCE OF NUMEROUS PSYCHOSOCIAL FACTORS, AT ITS CORE, DRUG ADDICTION INVOLVES A BIOLOGICAL PROCESS: the ability of repeated exposure to a drug of abuse to induce changes in a vulnerable brain that drive the compulsive seeking and taking of drugs, and loss of control over drug use, that define a state of addiction. ... A large body of literature has demonstrated that such ΔFosB induction in D1-type NAc neurons increases an animal's sensitivity to drug as well as natural rewards and promotes drug self-administration, presumably through a process of positive reinforcement
- ↑ Robison AJ, Nestler EJ (November 2011). "Transcriptional and epigenetic mechanisms of addiction". Nat. Rev. Neurosci. 12 (11): 623–637. doi:10.1038/nrn3111. PMC 3272277. PMID 21989194.
ΔFosB has been linked directly to several addiction-related behaviors ... Importantly, genetic or viral overexpression of ΔJunD, a dominant negative mutant of JunD which antagonizes ΔFosB- and other AP-1-mediated transcriptional activity, in the NAc or OFC blocks these key effects of drug exposure14,22–24. This indicates that ΔFosB is both necessary and sufficient for many of the changes wrought in the brain by chronic drug exposure. ΔFosB is also induced in D1-type NAc MSNs by chronic consumption of several natural rewards, including sucrose, high fat food, sex, wheel running, where it promotes that consumption14,26–30. This implicates ΔFosB in the regulation of natural rewards under normal conditions and perhaps during pathological addictive-like states. ... ΔFosB serves as one of the master control proteins governing this structural plasticity.
- ↑ Shin, J. Y.; Park, H. J.; Ahn, Y. H.; Lee, P. H. (2009). "Neuroprotective effect of l-dopa on dopaminergic neurons is comparable to pramipexol in MPTP-treated animal model of Parkinson's disease: a direct comparison study". Journal of Neurochemistry. 111 (4): 1042–50. doi:10.1111/j.1471-4159.2009.06381.x. PMID 19765187.
- ↑ Narahashi, T; Marszalec, W; Moriguchi, S; Yeh, JZ; Zhao, X (2003). "Unique mechanism of action of Alzheimer's drugs on brain nicotinic acetylcholine receptors and NMDA receptors". Life Sciences. 74 (2–3): 281–91. doi:10.1016/j.lfs.2003.09.015. PMID 14607256.
External links
 Media related to Neuropharmacology at Wikimedia Commons
Media related to Neuropharmacology at Wikimedia Commons
