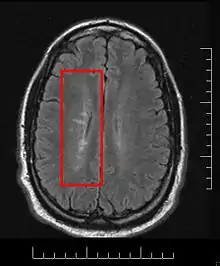
Multiple sclerosis and other demyelinating diseases of the central nervous system (CNS) produce lesions (demyelinated areas in the CNS) and glial scars or scleroses. They present different shapes and histological findings according to the underlying condition that produces them.
Demyelinating diseases are traditionally classified in two kinds: demyelinating myelinoclastic diseases and demyelinating leukodystrophic diseases. In the first group a normal and healthy myelin is destroyed by a toxic, chemical or autoimmune substance. In the second group, myelin is abnormal and degenerates.[1] The second group was denominated dysmyelinating diseases by Poser[2] Therefore, since Poser demyelinating diseases normally refers to the myelinoclastic part.
Demyelinating diseases of the CNS can be classified according to their pathogenesis into five non-exclusing categories: demyelination due to inflammatory processes, viral demyelination, demyelination caused by acquired metabolic derangements, hypoxic–ischaemic forms of demyelination and demyelination caused by focal compression.[3]
Non inflammatory demyelination
The four non-inflammatory possibilities are:
- viral demyelination,
- metabolic demyelination (Leukodystrophy and its sub-conditions, Adrenoleukodystrophy and Adrenomyeloneuropathy ),
- hypoxic–ischaemic forms of demyelination (Susac's syndrome, leukoaraiosis) and,
- demyelination caused by focal compression.
All these four types of demyelination are non-inflammatory and different to MS[4] even if some leukoencephalopathies can produce similar lesions[5]
Lesions produced by CNS Inflammatory Demyelinating diseases (IDS)
Typical lesions are similar to those of MS, but there are four kinds of atypical inflammatory demyelinating lesions: Ring-like (antibody-mediated), megacystic (tumefactive), Balo-like, and diffusely-infiltrating lesions.[6]
The list of the diseases that produce CNS demyelinating lesions is not complete, but it includes:
- Standard multiple sclerosis, the most known and extended variant.
- Devic's disease and neuromyelitis optica (NMO) (sometimes previously called optic-spinal MS)
- Acute disseminated encephalomyelitis or ADEM, a closely related disorder in which a known virus or vaccine triggers autoimmunity against myelin.
- Acute hemorrhagic leukoencephalitis, possibly a variant of Acute disseminated encephalomyelitis
- Balo concentric sclerosis, an unusual presentation of plaques forming concentric circles, which can sometimes get better spontaneously.
- Schilder disease or diffuse myelinoclastic sclerosis: is a rare disease that presents clinically as a pseudotumoural demyelinating lesion; and is more common in children.[7][8]
- Marburg multiple sclerosis, an aggressive form, also known as malignant, fulminant or acute MS.
- Tumefactive Multiple sclerosis: lesions whose size is more than 2 cm, with mass effect, oedema and/or ring enhancement[9][10]
- AntiMOG associated encephalomyelitis: Lesions similar to ADEM sometimes and to NMO some others. It is not normal, but can also appear like MS even with biopsy. These cases resemble MS pattern-II lesions.[11] The demyelinating lesion presents T-cells and macrophages around blood vessels, with preservation of oligodendrocytes and signs of complement system activation.
Confluent vs. perivenous demyelination
A special characteristic that makes a difference between MS and the several kinds of ADEM is the structure of the lesions, being strictly perivenous in ADEM and showing a confluence around veins in MS. Given that ADEM can be multiphase sometimes and MS can appear in children, this characteristic is often considered as the line that separates both conditions.[12]
The most typical of perivenous demyelination is ADEM
ADEM demyelination
ADEM can present plaque-like lesions which are indistinguishable from MS[13] Nevertheless, ADEM White Matter appears intact under Magnetization Transfer MRI, while MS shows problems (See NAWM).[14] Besides ADEM does not present "black holes" under MRI (zones with axonal damage) and lesions develop strictly around veins instead of the more relaxed rule for MS[15]
NMO demyelination
As with MS, several patterns have been described inside NMO, but they are heterogeneous inside the same individual, reflecting stages in the lesion evolution:[16]
- The first reflects complement deposition at the surface of astrocytes, associated with granulocyte infiltration and astrocyte necrosis
- demyelination, global tissue destruction and the formation of cystic, necrotic lesions (lesion type 2).
- Wallerian degeneration in lesion-related tracts (lesion type 3).
- Around active NMO lesions AQP4 may selectively be lost in the absence of aquaporin 1 (AQP1) loss or other structural damage (lesion type 4).
- Another pattern is characterized by clasmatodendrosis of astrocytes, defined by cytoplasmic swelling and vacuolation, beading and dissolution of their processes and nuclear alterations resembling apoptosis, which was associated with internalization of AQP4 and AQP1 and astrocyte apoptosis in the absence of complement activation. Such lesions give rise to extensive astrocyte loss, which may occur in part in the absence of any other tissue injury, such as demyelination or axonal degeneration (lesion type 5).
- Finally, lesions with a variable degree of astrocyte clasmatodendrosis are found, which show plaque-like primary demyelination that is associated with oligodendrocyte apoptosis, but with preservation of axons (lesion type 6).
Early active demyelinating NMO lesions may show complement within macrophages and oligodendrocyte apoptosis associated with a selective loss of minor myelin proteins, in addition to typical NMO features in a subset of active demyelinating NMO lesions[17]
Confluent demyelination

The demyelination around a vein is normally called "plaque". In MS plaques are reported to appear by coalescence of several confluent smaller demyelinations.
MS lesions
Normally MS lesions are small ovoid lesions, less than 2 cm. long, oriented perpendicular to the long axis of the brain's ventricles [18] Often they are disposed surrounding a vein[19]

Active and pre-active lesions appear as hyperintense areas under T2-weighted MRI. Pre-active lesion here refers to lesions localized in the normal appearing white matter, without apparent loss of myelin but nevertheless showing a variable degree of oedema, small clusters of microglial cells with enhanced major histocompatibility complex class II antigen, CD45 and CD68 antigen expression and a variable number of perivascular lymphocytes around small blood vessels[20]
Using high field MRI system, with several variants several areas show lesions, and can be spacially classified in infratentorial, callosal, juxtacortical, periventricular, and other white matter areas.[21] Other authors simplify this in three regions: intracortical, mixed gray-white matter, and juxtacortical.[22] Others classify them as hippocampal, cortical, and WM lesions,[23] and finally, others give seven areas: intracortical, mixed white matter-gray matter, juxtacortical, deep gray matter, periventricular white matter, deep white matter, and infratentorial lesions.[24] The distribution of the lesions could be linked to the clinical evolution[25]
Post-mortem autopsies reveal that gray matter demyelination occurs in the motor cortex, cingulate gyrus, cerebellum, thalamus and spinal cord.[26] Cortical lesions have been observed specially in people with SPMS but they also appear in RRMS and clinically isolated syndrome. They are more frequent in men than in women[27] and they can partly explain cognitive deficits.
It is known that two parameters of the cortical lesions, fractional anisotropy (FA) and mean diffusivity (MD), are higher in patients than in controls.[28] They are larger in SPMS than in RRMS and most of them remain unchanged for short follow-up periods. They do not spread into the subcortical white matter and never show gadolinium enhancement. Over a one-year period, CLs can increase their number and size in a relevant proportion of MS patients, without spreading into the subcortical white matter or showing inflammatory features similar to those of white matter lesions.[29]
The first plausible explanation of their distribution was published by Dr. Schelling. He said:
- The specific brain plaques of multiple sclerosis can only be caused by energetic venous back-jets set in motion by intermittent rises in the pressure in the large collecting veins of the neck, but especially of the chest..[30]
But no problems with chest veins was ever found.
This morphologic appearance was named Dawson's fingers by Charles Lumsden, after the Scottish pathologist James Walker Dawson,[31] who first defined the condition in 1916.
Dawson's fingers
"Dawson's fingers" is the name for the lesions around the ventricle-based brain veins[32][33] of patients with multiple sclerosis and antiMOG associated encephalomyelitis[34]
Though once thought to be specific of MS, it is known not to be the case.[35]
The condition is thought to be the result of inflammation or mechanical damage by blood pressure[30] around long axis of medular veins.
Dawson's fingers spread along, and from, large periventricular collecting veins, and are attributed to perivenular inflammation.[36]
Lesions far away from these veins are known as Steiner's splashes.[30]
Sometimes experimental autoimmune encephalomyelitis has been triggered in humans by accident or medical mistake. The damage in these cases fulfils all the pathological diagnostic criteria of MS and can therefore be classified as MS in its own right. The lesions were classified as pattern II in the Lucchinetti system. This case of human EAE also showed Dawson fingers.[37]
Tumefactive demyelinating lesions
Demyelinating lesions whose size is larger than 2 cm. They normally appear together with normal MS lesions, situation described as tumefactive multiple sclerosis. When they appear alone, they are usually named "Solitary sclerosis",[38] being more difficult to diagnose.
They look like intracranial neoplasms, and sometimes they get biopsied as suspected tumors. Proton MR spectroscopy can help in their diagnosis.[39]
Demyelination process in MS

The hallmark of MS is the lesion, which appears mainly in the white matter and shows macrophage mediated demyelination, BBB breakdown, inflammation and axon transection.
NAWM development
Demyelinating lesions begin with the appearance of some areas named NAWM (normal appearing white matter) which despite its name, is abnormal in several parameters. These areas show axonal transections and stressed oligodendrocytes (the cells responsible for maintaining the myelin), and randomly, they show clusters of activated microglia named pre-active lesions. These pre-lesions normally resolve themselves, though sometimes they spread towards a capilar vein.
BBB breakdown
This is followed by the blood–brain barrier (BBB) breakdown. BBB is a tight vascular barrier between the blood and brain that should prevent the passage of antibodies through it, but in MS patients it does not work. For unknown reasons special areas appear in the brain and spine, followed by leaks in the blood–brain barrier where immune cells infiltrate. This leads to the entrance of macrophages into the CNS, triggering the beginning of an immune-mediated attack against myelin. Gadolinium cannot cross a normal BBB and, therefore, Gadolinium-enhanced MRI is used to show BBB breakdowns.
Immune mediated attack
After the BBB breakdown, the immune-mediated attack against myelin happens. T cells, are a kind of lymphocyte that plays an important role in the body's defenses. The T cells recognize myelin as foreign and attack it, explaining why these cells are also called "autoreactive lymphocytes". Demyelination, further inflammation and axonal transection are the result.
The attack of myelin starts inflammatory processes, which triggers other immune cells and the release of soluble factors like cytokines and antibodies. Further breakdown of the blood–brain barrier, in turn cause a number of other damaging effects such as swelling, activation of macrophages, and more activation of cytokines and other destructive proteins.
Astrocytes can heal partially the lesion leaving a scar. These scars (sclerae) are the known plaques or lesions usually reported in MS. A repair process, called remyelination, takes place in early phases of the disease, but the oligodendrocytes are unable to completely rebuild the cell's myelin sheath. Repeated attacks lead to successively less effective remyelinations, until a scar-like plaque is built up around the damaged axons.
According to the view of most researchers, a special subset of lymphocytes, called T helper cells, specifically Th1 and Th17,[40] play a key role in the development of the lesion. Under normal circumstances, these lymphocytes can distinguish between self and non-self. However, in a person with MS, these cells recognize healthy parts of the central nervous system as foreign and attack them as if they were an invading virus, triggering inflammatory processes and stimulating other immune cells and soluble factors like cytokines and antibodies. Many of the myelin-recognizing T cells belong to a terminally differentiated subset called co-stimulation-independent effector-memory T cells.[41][42][43][44][45][46][47][48][49][50][51] Recently other type of immune cells, B Cells, have been also implicated in the pathogenesis of MS[52] and in the degeneration of the axons.[53]
The axons themselves can also be damaged by the attacks.[54] Often, the brain is able to compensate for some of this damage, due to an ability called neuroplasticity. MS symptoms develop as the cumulative result of multiple lesions in the brain and spinal cord. This is why symptoms can vary greatly between different individuals, depending on where their lesions occur.
Lesion recovery
Under laboratory conditions, stem cells are quite capable of proliferating and differentiating into remyelinating oligodendrocytes; it is therefore suspected that inflammatory conditions or axonal damage somehow inhibit stem cell proliferation and differentiation in affected areas[55] It is possible to predict how much and when lesion will recover[56]
Related to this, it was found in 2016 that neural cells of primary progressive patients (PPMS) do have some kind of problem to protect neuroprotection against demyelination or oligodendrocytes, compared to healthy subjects. Some genetics seem to underlie the problem as this was shown using Induced pluripotent stem cell (iPSC) as neural progenitor cells (NPC)[57]
Demyelination patterns in standard MS
Four different damage patterns, known as Lassmann patterns,[58] have been identified by her team in the scars of the brain tissue in multiple sclerosis, and they are used sometimes as a basis for describing lesions in other demyelinating diseases.
- Pattern I
- The scar presents T-cells and macrophages around blood vessels, with preservation of oligodendrocytes, but no signs of complement system activation.[59]
- Pattern II
- The scar presents T-cells and macrophages around blood vessels, with preservation of oligodendrocytes, as before, but also signs of complement system activation can be found.[60] Though this pattern could be considered similar to damage seen in NMO, some authors report no AQP4 damage in pattern II lesions[61]
- Pattern III
- The scars are diffuse with inflammation, distal oligodendrogliopathy and microglial activation. There is also loss of myelin-associated glycoprotein (MAG). The scars do not surround the blood vessels, and in fact, a rim of preserved myelin appears around the vessels. There is evidence of partial remyelinization and oligodendrocyte apoptosis. For some researchers this pattern is an early stage of the evolution of the others.[62]
- Pattern IV
- The scar presents sharp borders and oligodendrocyte degeneration, with a rim of normal appearing white matter. There is a lack of oligodendrocytes in the center of the scar. There is no complement activation or MAG loss.
The meaning of this fact is controversial. For some investigation teams it means that MS is a heterogeneous disease. Others maintain that the shape of the scars can change with time from one type to other and this could be a marker of the disease evolution.[63] Anyway, the heterogeneity could be true only for the early stage of the disease.[64] Some lesions present mitochondrial defects that could distinguish types of lesions.[65] Currently antibodies to lipids and peptides in sera, detected by microarrays, can be used as markers of the pathological subtype given by brain biopsy.[66]
After some debate among research groups, currently the heterogeneity hypothesis looks like accepted[17]
See also
External links
Sources
- ↑ Fernández O.; Fernández V.E.; Guerrero M. (2015). "Demyelinating diseases of the central nervous system". Medicine. 11 (77): 4601–4609. doi:10.1016/j.med.2015.04.001.
- ↑ Poser C. M. (1961). "Leukodystrophy and the Concept of Dysmyelination". Arch Neurol. 4 (3): 323–332. doi:10.1001/archneur.1961.00450090089013. PMID 13737358.
- ↑ Love S (2006). "Demyelinating diseases". J Clin Pathol. 59 (11): 1151–1159. doi:10.1136/jcp.2005.031195. PMC 1860500. PMID 17071802.
- ↑ John R. Hesselink, MD, FACR. DEMYELINATING DISEASES OF THE BRAIN
- ↑ Gray F, Chimelli L, Mohr M, Clavelou P, Scaravilli F, Poirier J (1991). "Fulminating multiple sclerosis‐like leukoencephalopathy revealing human immunodeficiency virus infection". Neurology. 41 (1): 105–9. doi:10.1212/WNL.41.1.105. PMID 1985273. S2CID 5541633.
- ↑ Seewann A, et al. (2008). "MRI characteristics of atypical idiopathic inflammatory demyelinating lesions of the brain". Journal of Neurology. 255 (1): 1–10. doi:10.1007/s00415-007-0754-x. PMID 18004634. S2CID 1411872.
- ↑ Garrido C, Levy-Gomes A, Teixeira J, Temudo T (2004). "[Schilder's disease: two new cases and a review of the literature]". Revista de Neurología (in Spanish). 39 (8): 734–8. doi:10.33588/rn.3908.2003023. PMID 15514902.
- ↑ Afifi AK, Bell WE, Menezes AH, Moore SA (1994). "Myelinoclastic diffuse sclerosis (Schilder's disease): report of a case and review of the literature". J. Child Neurol. 9 (4): 398–403. CiteSeerX 10.1.1.1007.559. doi:10.1177/088307389400900412. PMID 7822732. S2CID 38765870.
- ↑ Lucchinetti CF, Gavrilova RH, Metz I, et al. (July 2008). "Clinical and radiographic spectrum of pathologically confirmed tumefactive multiple sclerosis". Brain. 131 (Pt 7): 1759–75. doi:10.1093/brain/awn098. PMC 2442427. PMID 18535080.
- ↑ Given CA, Stevens BS, Lee C (1 January 2004). "The MRI appearance of tumefactive demyelinating lesions". AJR Am J Roentgenol. 182 (1): 195–9. doi:10.2214/ajr.182.1.1820195. PMID 14684539.
- ↑ Spadaro Melania; et al. (2015). "Histopathology and clinical course of MOG-antibody-associated encephalomyelitis". Annals of Clinical and Translational Neurology. 2 (3): 295–301. doi:10.1002/acn3.164. PMC 4369279. PMID 25815356.
- ↑ Young NP, Weinshenker BG, Parisi JE, Scheithauer B, Giannini C, Roemer SF, Thomsen KM, Mandrekar JN, Erickson BJ, Lucchinetti CF (2010). "Perivenous demyelination: association with clinically defined acute disseminated encephalomyelitis and comparison with pathologically confirmed multiple sclerosis". Brain. 133 (2): 333–348. doi:10.1093/brain/awp321. PMC 2822631. PMID 20129932.
- ↑ Guenther AD, Munoz DG (2013). "b Plaque-like demyelination in acute disseminated encephalomyelitis (ADEM) - an autopsy case report". Clin Neuropathol. 32 (6): 486–91. doi:10.5414/NP300634. PMID 23863345.
- ↑ Book, Clinical Neuroimmunology: Multiple Sclerosis and Related Disorders, Syed A. Rizvi, Patricia K. Coyle, Springer Science & Business Media, 3 sept. 2011 - 404 pages
- ↑ Young Nathan P (Feb 2008). "D.O Acute Disseminated Encephalomyelitis: Current Understanding and Controversies" (PDF). Semin Neurol. 28 (1): 84–94. doi:10.1055/s-2007-1019130. PMID 18256989.
- ↑ Misu T, Höftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S, Nakashima I, Konno H, Bradl M, Garzuly F, Itoyama Y, Aoki M, Lassmann H (Jun 2013). "Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica". Acta Neuropathol. 125 (6): 815–27. doi:10.1007/s00401-013-1116-7. PMC 3661909. PMID 23579868.
- 1 2 Brück W, Popescu B, Lucchinetti CF, Markovic-Plese S, Gold R, Thal DR, Metz I (Sep 2012). "Neuromyelitis optica lesions may inform multiple sclerosis heterogeneity debate". Annals of Neurology. 72 (3): 385–94. doi:10.1002/ana.23621. PMID 23034911. S2CID 1662420.
- ↑ Lucchinetti C. F.; et al. (2008). "Clinical and radiographic spectrum of pathologically confirmed tumefactive multiple sclerosis". Brain. 131 (Pt 7): 1759–1775. doi:10.1093/brain/awn098. PMC 2442427. PMID 18535080.
- ↑ Barnett Michael H.; Sutton Ian (2006). "The pathology of multiple sclerosis: a paradigm shift". Curr Opin Neurol. 19 (3): 242–247. doi:10.1097/01.wco.0000227032.47458.cb. PMID 16702829. S2CID 2687650.
- ↑ De Groot CJ, Bergers E, Kamphorst W, Ravid R, Polman CH, Barkhof F, van der Valk P (2001). "Post-mortem MRI-guided sampling of multiple sclerosis brain lesions increased yield of active demyelinating and (pre)active lesions". Brain. 124 (8): 1635–1645. doi:10.1093/brain/124.8.1635. PMID 11459754.
- ↑ Wattjes MP, Harzheim M, Kuhl CK, et al. (1 September 2006). "Does high-field MR imaging have an influence on the classification of patients with clinically isolated syndromes according to current diagnostic mr imaging criteria for multiple sclerosis?". AJNR Am J Neuroradiol. 27 (8): 1794–8. PMC 8139807. PMID 16971638.
- ↑ Nelson F, Poonawalla AH, Hou P, Huang F, Wolinsky JS, Narayana PA (October 2007). "Improved identification of intracortical lesions in multiple sclerosis with phase-sensitive inversion recovery in combination with fast double inversion recovery MR imaging". AJNR Am J Neuroradiol. 28 (9): 1645–9. doi:10.3174/ajnr.A0645. PMC 8134176. PMID 17885241.
- ↑ Roosendaal SD, Moraal B, Vrenken H, et al. (April 2008). "In vivo MR imaging of hippocampal lesions in multiple sclerosis". J Magn Reson Imaging. 27 (4): 726–31. doi:10.1002/jmri.21294. PMID 18302199. S2CID 46567107.
- ↑ Geurts JJ, Pouwels PJ, Uitdehaag BM, Polman CH, Barkhof F, Castelijns JA (July 2005). "Intracortical lesions in multiple sclerosis: improved detection with 3D double inversion-recovery MR imaging". Radiology. 236 (1): 254–60. doi:10.1148/radiol.2361040450. PMID 15987979.
- ↑ Sampat MP, Berger AM, Healy BC, et al. (October 2009). "Regional white matter atrophy--based classification of multiple sclerosis in cross-sectional and longitudinal data". AJNR Am J Neuroradiol. 30 (9): 1731–9. doi:10.3174/ajnr.A1659. PMC 2821733. PMID 19696139.
- ↑ Gilmore CP, Donaldson I, Bö L, Owens T, Lowe JS, Evangelou N (October 2008). "Regional variations in the extent and pattern of grey matter demyelination in Multiple Sclerosis: a comparison between the cerebral cortex, cerebellar cortex, deep grey matter nuclei and the spinal cord". J Neurol Neurosurg Psychiatry. 80 (2): 182–7. doi:10.1136/jnnp.2008.148767. hdl:1871/22404. PMID 18829630. S2CID 7545552.
- ↑ Calabrese M, De Stefano N, Atzori M, et al. (2007). "Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis". Arch. Neurol. 64 (10): 1416–22. doi:10.1001/archneur.64.10.1416. PMID 17923625.
- ↑ Poonawalla AH, Hasan KM, Gupta RK, et al. (2008). "Diffusion-Tensor MR Imaging of Cortical Lesions in Multiple Sclerosis: Initial Findings". Radiology. 246 (3): 880–6. doi:10.1148/radiol.2463070486. PMID 18195384.
- ↑ Calabrese M, Filippi M, Rovaris M, Mattisi I, Bernardi V, Atzori M, Favaretto A, Barachino L, Rinaldi L, Romualdi C, Perini P, Gallo P (2008). "Morphology and evolution of cortical lesions in multiple sclerosis. A longitudinal MRI study". NeuroImage. 42 (4): 1324–8. doi:10.1016/j.neuroimage.2008.06.028. PMID 18652903. S2CID 29732090.
- 1 2 3 Schelling F. MS: The image and its message
- ↑ Dawson J.W. (1916). "The histology of disseminated sclerosis". Trans. R. Soc. Edinb. 50 (3): 517–740. doi:10.1017/s0080456800027174. S2CID 75975878.
- ↑ Peter Reimer, Paul M. Parizel, Falko-Alexander Stichnoth. Clinical MR imaging: a practical approach.
- ↑ Dawson fingers, at Radiopedia
- ↑ Which Brain Lesion Locations Differentiate Multiple Sclerosis (MS) from Neuromyelitis Optica Spectrum Disorders (NMOSD) and MOG Antibody Disorder (MOGAD)? (2399) Jasmin Patel, Antonio Pires, Anna Derman, Erik Charlson, Girish Fatterpekar, Ilya Kister, Neurology Apr 2020, 94 (15 Supplement) 2399;
- ↑ Del Brutto, Oscar H.; Mera, Robertino M.; Costa, Aldo F.; Silva, Patricia; Del Brutto, Victor J. (17 September 2020). "Dawson Fingers in Older Adults with Cerebral Small Vessel Disease: A Population Study". European Neurology. 83 (4): 421–425. doi:10.1159/000510076. PMID 32942284. S2CID 221789043.
- ↑ Suzanne Palmer, William G. Bradley, Dar-Yeong Chen, Sangita Patel, Subcallosal Striations: Early Findings of Multiple Sclerosis on Sagittal, Thin-Section, Fast FLAIR MR Images
- ↑ Höftberger R, Leisser M, Bauer J, Lassmann H (Dec 2015). "Autoimmune encephalitis in humans: how closely does it reflect multiple sclerosis?". Acta Neuropathol Commun. 3 (1): 80. doi:10.1186/s40478-015-0260-9. PMC 4670499. PMID 26637427.
- ↑ Kepes JJ (2004). "Large focal tumor-like demyelinating lesions of the brain: Intermediate entity between multiple sclerosis and acute disseminated encephalomyelitis? A study of 31 patients". Annals of Neurology. 33 (1): 18–27. doi:10.1002/ana.410330105. PMID 8494332. S2CID 43813581.
- ↑ Saindane; et al. (2002). "Proton MR Spectroscopy of Tumefactive Demyelinating Lesions". American Journal of Neuroradiology. 23 (8): 1378–1386. PMC 7976261. PMID 12223381.
- ↑ Fransson ME, Liljenfeldt LS, Fagius J, Tötterman TH, Loskog AS (2009). "The T-cell pool is anergized in patients with multiple sclerosis in remission". Immunology. 126 (1): 92–101. doi:10.1111/j.1365-2567.2008.02881.x. PMC 2632699. PMID 18624727.
- ↑ Markovic-Plese S, Cortese I, Wandinger KP, McFarland HF, Martin R (October 2001). "CD4+CD28- costimulation-independent T cells in multiple sclerosis". J. Clin. Invest. 108 (8): 1185–94. doi:10.1172/JCI12516. PMC 209525. PMID 11602626.
- ↑ Wulff H, Calabresi PA, Allie R, et al. (June 2003). "The voltage-gated Kv1.3 K(+) channel in effector memory T cells as new target for MS". J. Clin. Invest. 111 (11): 1703–13. doi:10.1172/JCI16921. PMC 156104. PMID 12782673.
- ↑ Rus H, Pardo CA, Hu L, et al. (August 2005). "The voltage-gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain". Proc. Natl. Acad. Sci. U.S.A. 102 (31): 11094–9. Bibcode:2005PNAS..10211094R. doi:10.1073/pnas.0501770102. PMC 1182417. PMID 16043714.
- ↑ Beeton C, Chandy KG (December 2005). "Potassium channels, memory T cells, and multiple sclerosis". Neuroscientist. 11 (6): 550–62. doi:10.1177/1073858405278016. PMID 16282596. S2CID 24416951.
- ↑ Okuda Y, Okuda M, Apatoff BR, Posnett DN (August 2005). "The activation of memory CD4(+) T cells and CD8(+) T cells in patients with multiple sclerosis". J. Neurol. Sci. 235 (1–2): 11–7. doi:10.1016/j.jns.2005.02.013. PMID 15972217. S2CID 25020233.
- ↑ Krakauer M, Sorensen PS, Sellebjerg F (December 2006). "CD4(+) memory T cells with high CD26 surface expression are enriched for Th1 markers and correlate with clinical severity of multiple sclerosis". J. Neuroimmunol. 181 (1–2): 157–64. doi:10.1016/j.jneuroim.2006.09.006. PMID 17081623. S2CID 14134201.
- ↑ Ratts RB, Karandikar NJ, Hussain RZ, et al. (September 2006). "Phenotypic characterization of autoreactive T cells in multiple sclerosis". J. Neuroimmunol. 178 (1–2): 100–10. doi:10.1016/j.jneuroim.2006.06.010. PMID 16901549. S2CID 7516665.
- ↑ Haegele KF, Stueckle CA, Malin JP, Sindern E (February 2007). "Increase of CD8+ T-effector memory cells in peripheral blood of patients with relapsing-remitting multiple sclerosis compared to healthy controls". J. Neuroimmunol. 183 (1–2): 168–74. doi:10.1016/j.jneuroim.2006.09.008. PMID 17084910. S2CID 41262535.
- ↑ Jilek S, Schluep M, Rossetti AO, et al. (April 2007). "CSF enrichment of highly differentiated CD8+ T cells in early multiple sclerosis". Clin. Immunol. 123 (1): 105–13. doi:10.1016/j.clim.2006.11.004. PMID 17188575.
- ↑ Miyazaki Y, Iwabuchi K, Kikuchi S, et al. (September 2008). "Expansion of CD4+CD28- T cells producing high levels of interferon-{gamma} in peripheral blood of patients with multiple sclerosis". Mult. Scler. 14 (8): 1044–55. doi:10.1177/1352458508092809. PMID 18573819. S2CID 12786590.
- ↑ Lünemann JD, Jelcić I, Roberts S, Lutterotti A, Tackenberg B, Martin R, Münz C (August 2008). "EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2". The Journal of Experimental Medicine. 205 (8): 1763–73. doi:10.1084/jem.20072397. PMC 2525578. PMID 18663124.
- ↑ Hauser SL, Waubant E, Arnold DL, et al. (February 2008). "B-cell depletion with rituximab in relapsing-remitting multiple sclerosis". N Engl J Med. 358 (7): 676–88. doi:10.1056/NEJMoa0706383. PMID 18272891.
- ↑ Cause of nerve fiber damage in multiple sclerosis identified
- ↑ Pascual AM, Martínez-Bisbal MC, Boscá I, et al. (2007). "Axonal loss is progressive and partly dissociated from lesion load in early multiple sclerosis". Neurology. 69 (1): 63–7. doi:10.1212/01.wnl.0000265054.08610.12. PMID 17606882. S2CID 23230073.
- ↑ Wolswijk G (15 January 1998). "Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells". J. Neurosci. 18 (2): 601–9. doi:10.1523/JNEUROSCI.18-02-00601.1998. PMC 6792542. PMID 9425002.
- ↑ Jordan D. Dworkina el al. PREVAIL: Predicting Recovery through Estimation and Visualization of Active and Incident Lesions. NeuroImage: Clinical, 2 August 2016
- ↑ Nicaise AM, Banda E, Guzzo RM, Russomanno K, Castro-Borrero W, Willis CM, Johnson KM, Lo AC, Crocker SJ (2017). "iPS-derived neural progenitor cells from PPMS patients reveal defect in myelin injury response". Exp Neurol. 288: 114–121. doi:10.1016/j.expneurol.2016.11.012. PMID 27865736. S2CID 25254295.
- ↑ Gold R, Linington C (July 2002). "Devic's disease: bridging the gap between laboratory and clinic". Brain. 125 (Pt 7): 1425–7. doi:10.1093/brain/awf147. PMID 12076994.
- ↑ Holmes, Nick (15 November 2001). "Part 1B Pathology: Lecture 11 - The Complement System". Archived from the original on 9 January 2006. Retrieved 2006-05-10.
- ↑ Lucchinetti, Claudia; Wolfgang Brück; Joseph Parisi; Bernd Scheithauer; Moses Rodriguez; Hans Lassmann (December 1999). "A quantitative analysis of oligodendrocytes in multiple sclerosis lesions - A study of 113 cases". Brain. 122 (12): 2279–2295. doi:10.1093/brain/122.12.2279. PMID 10581222.
- ↑ Kale N, Pittock SJ, Lennon VA, et al. (October 2009). "Humoral pattern II multiple sclerosis pathology not associated with neuromyelitis Optica IgG". Arch. Neurol. 66 (10): 1298–9. doi:10.1001/archneurol.2009.199. PMC 2767176. PMID 19822791.
- ↑ Barnett MH, Prineas JW (April 2004). "Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion" (PDF). Annals of Neurology. 55 (4): 458–68. doi:10.1002/ana.20016. PMID 15048884. S2CID 5659495. Archived from the original (PDF) on 2013-10-29. Retrieved 2013-06-02.
- ↑ Michael H. Barnett, MBBS; John W. Prineas, MBBS (2004). "Relapsing and Remitting Multiple Sclerosis: Pathology of the Newly Forming Lesion" (PDF). Annals of Neurology. 55 (1): 458–468. doi:10.1002/ana.20016. PMID 15048884. S2CID 5659495.
- ↑ Breij EC, Brink BP, Veerhuis R, et al. (2008). "Homogeneity of active demyelinating lesions in established multiple sclerosis". Annals of Neurology. 63 (1): 16–25. doi:10.1002/ana.21311. PMID 18232012. S2CID 205340842.
- ↑ Mahad D, Ziabreva I, Lassmann H, Turnbull D (2008). "Mitochondrial defects in acute multiple sclerosis lesions". Brain: A Journal of Neurology. 131 (Pt 7): 1722–35. doi:10.1093/brain/awn105. PMC 2442422. PMID 18515320.
- ↑ Quintana FJ, Farez MF, Viglietta V, et al. (December 2008). "Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis". Proc. Natl. Acad. Sci. U.S.A. 105 (48): 18889–94. Bibcode:2008PNAS..10518889Q. doi:10.1073/pnas.0806310105. PMC 2596207. PMID 19028871.