
A molecular solid is a solid consisting of discrete molecules. The cohesive forces that bind the molecules together are van der Waals forces, dipole-dipole interactions, quadrupole interactions, π-π interactions, hydrogen bonding, halogen bonding, London dispersion forces, and in some molecular solids, coulombic interactions.[3][4][5][6][7][8][9][10] Van der Waals, dipole interactions, quadrupole interactions, π-π interactions, hydrogen bonding, and halogen bonding (2-127 kJ mol−1)[10] are typically much weaker than the forces holding together other solids: metallic (metallic bonding, 400-500 kJ mol−1),[4] ionic (Coulomb’s forces, 700-900 kJ mol−1),[4] and network solids (covalent bonds, 150-900 kJ mol−1).[4][10] Intermolecular interactions, typically do not involve delocalized electrons, unlike metallic and certain covalent bonds. Exceptions are charge-transfer complexes such as the tetrathiafulvane-tetracyanoquinodimethane (TTF-TCNQ), a radical ion salt.[5] These differences in the strength of force (i.e. covalent vs. van der Waals) and electronic characteristics (i.e. delocalized electrons) from other types of solids give rise to the unique mechanical, electronic, and thermal properties of molecular solids.[3][4][5][8]
Molecular solids are poor electrical conductors,[4][5] although some, such as TTF-TCNQ are semiconductors (ρ = 5 x 102 Ω−1 cm−1).[5] They are still substantially less than the conductivity of copper (ρ = 6 x 105 Ω−1 cm−1).[8] Molecular solids tend to have lower fracture toughness (sucrose, KIc = 0.08 MPa m1/2)[11] than metal (iron, KIc = 50 MPa m1/2),[11] ionic (sodium chloride, KIc = 0.5 MPa m1/2),[11] and covalent solids (diamond, KIc = 5 MPa m1/2).[12] Molecular solids have low melting (Tm) and boiling (Tb) points compared to metal (iron), ionic (sodium chloride), and covalent solids (diamond).[4][5][8][13] Examples of molecular solids with low melting and boiling temperatures include argon, water, naphthalene, nicotine, and caffeine (see table below).[13][14] The constituents of molecular solids range in size from condensed monatomic gases[15] to small molecules (i.e. naphthalene and water)[16][17] to large molecules with tens of atoms (i.e. fullerene with 60 carbon atoms).[18]
| Type of Solid | Material | Tm (°C) | Tb (°C) |
|---|---|---|---|
| Metallic | Iron | 1,538[13] | 2,861[13] |
| Ionic | Sodium chloride | 801[13] | 1,465[13] |
| Covalent | Diamond | 4,440[13] | - |
| Molecular | Argon | -189.3[13] | -185.9[13] |
| Molecular | Water | 0[13] | 100[13] |
| Molecular | Naphthalene | 80.1[13] | 217.9[13] |
| Molecular | Nicotine | -79[13] | 491[13] |
| Molecular | Caffeine | 235.6[13] | 519.9[14] |
Composition and structure
Molecular solids may consist of single atoms, diatomic, and/or polyatomic molecules.[1][2][3][4][5][6][7] The intermolecular interactions between the constituents dictate how the crystal lattice of the material is structured.[19][20][21] All atoms and molecules can partake in van der Waals and London dispersion forces (sterics). It is the lack or presence of other intermolecular interactions based on the atom or molecule that affords materials unique properties.[19]
Van der Waals forces
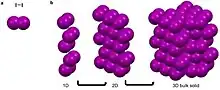
Argon, is a noble gas that has a full octet, no charge, and is nonpolar.[3][4][7][8] These characteristics make it unfavorable for argon to partake in metallic, covalent, and ionic bonds as well as most intermolecular interactions.[3][4][7][8] It can though partake in van der Waals and London dispersion forces.[3][4] These weak self-interactions are isotropic and result in the long-range ordering of the atoms into face centered cubic packing when cooled below -189.3.[13] Similarly iodine, a linear diatomic molecule has a net dipole of zero and can only partake in van der Waals interactions that are fairly isotropic.[3][4][7][8] This results in the bipyramidal symmetry.
Dipole-dipole and quadrupole interactions
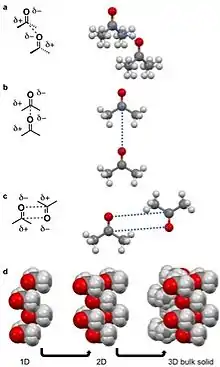
For acetone dipole-dipole interactions are a major driving force behind the structure of its crystal lattice. The negative dipole is caused by oxygen. Oxygen is more electronegative than carbon and hydrogen,[13] causing a partial negative (δ-) and positive charge (δ+) on the oxygen and remainder of the molecule, respectively.[3][5] The δ- orienttowards the δ+ causing the acetone molecules to prefer to align in a few configurations in a δ- to δ+ orientation (pictured left). The dipole-dipole and other intermolecular interactions align to minimize energy in the solid state and determine the crystal lattice structure.
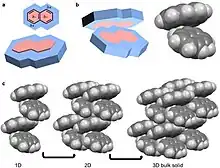
A quadrupole, like a dipole, is a permanent pole but the electric field of the molecule is not linear as in acetone, but in two dimensions.[25] Examples of molecular solids with quadrupoles are octafluoronaphthalene and naphthalene.[17][25] Naphthalene consists of two joined conjugated rings. The electronegativity of the atoms of this ring system and conjugation cause a ring current resulting in a quadrupole. For naphthalene, this quadrupole manifests in a δ- and δ+ accumulating within and outside the ring system, respectively.[4][5][6][10][25] Naphthalene assembles through the coordination of δ- of one molecules to the δ+ of another molecule.[4][5][6] This results in 1D columns of naphthalene in a herringbone configuration. These columns then stack into 2D layers and then 3D bulk materials. Octafluoronaphthalene follows this path of organization to build bulk material except the δ- and δ+ are on the exterior and interior of the ring system, respectively.[5]
Hydrogen and halogen bonding

A hydrogen bond is a specific dipole where a hydrogen atom has a partial positive charge (δ+) to due a neighboring electronegative atom or functional group.[9][10] Hydrogen bonds are amongst the strong intermolecular interactions know other than ion-dipole interactions.[10] For intermolecular hydrogen bonds the δ+ hydrogen interacts with a δ- on an adjacent molecule. Examples of molecular solids that hydrogen bond are water, amino acids, and acetic acid.[3][5][8][10] For acetic acid, the hydrogen (δ+) on the alcohol moiety of the carboxylic acid hydrogen bonds with other the carbonyl moiety (δ-) of the carboxylic on the adjacent molecule. This hydrogen bond leads a string of acetic acid molecules hydrogen bonding to minimize free energy.[10][26] These strings of acetic acid molecules then stack together to build solids.
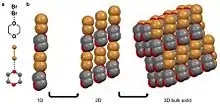
A halogen bond is when an electronegative halide participates in a noncovalent interaction with a less electronegative atom on an adjacent molecule.[10][28] Examples of molecular solids that halogen bond are hexachlorobenzene[11][29] and a cocrystal of bromine 1,4-dioxane.[27] For the second example, the δ- bromine atom in the diatomic bromine molecule is aligning with the less electronegative oxygen in the 1,4-dioxane. The oxygen in this case is viewed as δ+ compared to the bromine atom. This coordination results in a chain-like organization that stack into 2D and then 3D.[27]
Coulombic interactions
Coulombic interactions are manifested in some molecular solids. A well-studied example is the radical ion salt TTF-TCNQ with a conductivity of 5 x 102 Ω−1 cm−1,[5] much closer to copper (ρ = 6 x 105 Ω−1 cm−1)[8] than many molecular solids.[31][18][30] The coulombic interaction in TTF-TCNQ stems from the large partial negative charge (δ = -0.59) on the cyano- moiety on TCNQ at room temperature.[5] For reference, a completely charged molecule δ = ±1.[5] This partial negative charge leads to a strong interaction with the thio- moiety of the TTF. The strong interaction leads to favorable alignment of these functional groups adjacent to each other in the solid state.[5][30] While π-π interactions cause the TTF and TCNQ to stack in separate columns.[10][30]
Allotropes
One form of an element may be a molecular solid, but another form of that same element may not be a molecular solid.[3][4][5] For example, solid phosphorus can crystallize as different allotropes called "white", "red", and "black" phosphorus. White phosphorus forms molecular crystals composed of tetrahedral P4 molecules.[32] Heating at ambient pressure to 250 °C or exposing to sunlight converts white phosphorus to red phosphorus where the P4 tetrahedra are no longer isolated, but connected by covalent bonds into polymer-like chains.[33] Heating white phosphorus under high (GPa) pressures converts it to black phosphorus which has a layered, graphite-like structure.[34][35]
The structural transitions in phosphorus are reversible: upon releasing high pressure, black phosphorus gradually converts into the red phosphorus, and by vaporizing red phosphorus at 490 °C in an inert atmosphere and condensing the vapor, covalent red phosphorus can be transformed into the molecular solid, white phosphorus.[36]
 |
 |
 | |||
| White, red, violet, and black phosphorus samples | Structure unit of white phosphorus |
Structures of red | violet | and black phosphorus |
Similarly, yellow arsenic is a molecular solid composed of As4 units.[37] Some forms of sulfur and selenium are composed of S8 (or Se8) units and are molecular solids at ambient conditions, but converted into covalent allotropes having atomic chains extending throughout the crystal.[38][39]
Properties
Since molecular solids are held together by relatively weak forces they tend to have low melting and boiling points, low mechanical strength, low electrical conductivity, and poor thermal conductivity.[3][4][5][6][7][8]it will Also, depending on the structure of the molecule the intermolecular forces may have directionality leading to anisotropy of certain properties.[4][5][8]
Melting and boiling points
The characteristic melting point of metals and ionic solids is ~ 1000 °C and greater, while molecular solids typically melt closer to 300 °C (see table), thus many corresponding substances are either liquid (ice) or gaseous (oxygen) at room temperature.[4][6][7][8][40] This is due to the elements involved, the molecules they form, and the weak intermolecular interactions of the molecules.
| Formula | Tm °C |
|---|---|
| H2 | −259.1 |
| F2 | −219.6 |
| O2 | −218.8 |
| N2 | −210.0 |
| CH4 | −182.4 |
| C2H6 | −181.8 |
| C3H8 | −165.0 |
| C4H10 | −138.3 |
| C5H12 | −129.8 |
| Cl2 | −101.6 |
| C6H14 | −95.3 |
| HBr | −86.8 |
| HF | −80.0 |
| NH3 | −80.0 |
| HI | −50.8 |
| C10H22 | −29.7 |
| HCl | −27.3 |
| Br2 | −7.2 |
| H2O | 0.0 |
| C6H6 | 5.5 |
| I2 | 113.7 |
| S8 | 119.0 |
| C6Cl6 | 220.0 |
- See also
higher alkanes
Allotropes of phosphorus are useful to further demonstrate this structure-property relationship. White phosphorus, a molecular solid, has a relatively low density of 1.82 g/cm3 and melting point of 44.1 °C; it is a soft material which can be cut with a knife. When it is converted to the covalent red phosphorus, the density goes to 2.2–2.4 g/cm3 and melting point to 590 °C, and when white phosphorus is transformed into the (also covalent) black phosphorus, the density becomes 2.69–3.8 g/cm3 and melting temperature ~200 °C. Both red and black phosphorus forms are significantly harder than white phosphorus.[43]
Mechanical properties
Molecular solids can be either ductile or brittle, or a combination depending on the crystal face stressed.[5][11] Both ductile and brittle solids undergo elastic deformation till they reach the yield stress.[8][11] Once the yield stress is reached ductile solids undergo a period of plastic deformation, and eventually fracture. Brittle solids fracture promptly after passing the yield stress.[8][11] Due to the asymmetric structure of most molecules, many molecular solids have directional intermolecular forces.[11] This phenomenon can lead to anisotropic mechanical properties. Typically a molecular solid is ductile when it has directional intermolecular interactions. This allows for dislocation between layers of the crystal much like metals.[5][8][11]
One example of a ductile molecular solid, that can be bent 180°, is hexachlorobenzene (HCB).[11][29] In this example the π-π interactions between the benzene cores are stronger than the halogen interactions of the chlorides. This difference leads to its flexibility.[11][29] This flexibility is anisotropic; to bend HCB to 180° you must stress the [001] face of the crystal.[29] Another example of a flexible molecular solid is 2-(methylthio)nicotinic acid (MTN).[11][29] MTN is flexible due to its strong hydrogen bonding and π-π interactions creating a rigid set of dimers that dislocate along the alignment of their terminal methyls.[29] When stressed on the [010] face this crystal will bend 180°.[29] Note, not all ductile molecular solids bend 180° and some may have more than one bending faces.[29]
Electrical properties
Molecular solids are generally insulators.[5][18] This large band gap (compared to germanium at 0.7 eV)[8] is due to the weak intermolecular interactions, which result in low charge carrier mobility. Some molecular solids exhibit electrical conductivity, such as TTF-TCNQ with ρ = 5 x 102 Ω−1 cm−1 but in such cases orbital overlap is evident in the crystal structure. Fullerenes, which are insulating, become conducting or even superconducting upon doping.[44]
Thermal properties
Molecular solids have many thermal properties: specific heat capacity, thermal expansion, and thermal conductance to name a few.[3][5][6][7][8] These thermal properties are determined by the intra- and intermolecular vibrations of the atoms and molecules of the molecular solid. While transitions of an electron do contribute to thermal properties, their contribution is negligible compared to the vibrational contribution.[5][8]
See also
References
- 1 2 Simon, A.; Peters, K. (1980). "Single-Crystal Refinement of the Structure of Carbon Dioxide". Acta Crystallogr. B. 36 (11): 2750–2751. doi:10.1107/s0567740880009879.
- 1 2 Lehmann, C. W.; Stowasser, Frank (2007). "The Crystal Structure of Anhydrous Beta-Caffeine as Determined from X-ray Powder-Diffraction Data". Chemistry: A European Journal. 13 (10): 2908–2911. doi:10.1002/chem.200600973. PMID 17200930.
- 1 2 3 4 5 6 7 8 9 10 11 12 Hall, George (1965). Molecular Solid State Physics. Berlin, Germany: Springer-Verlag.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Fahlman, B. D. (2011). Materials Chemistry. Berlin, Germany: Springer.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Schwoerer, M.; Wolf, H. C. (2007). Organic Molecular Solids. Weinheim, Germany: Wiley-VCH.
- 1 2 3 4 5 6 7 Omar, M. A. (2002). Elementary Solid State Physics. London, England: Pearson.
- 1 2 3 4 5 6 7 8 Patterson, J.; Bailey, B. (2010). Solid-State Physics. Berlin, Germany: Springer.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Turton, R. (2010). The Physics of Solids. New York, New York: Oxford University Press Inc.
- 1 2 Keer, H. V. (1993). Principles of Solid State. Hoboken, New Jersey: Wiley Eastern Limited.
- 1 2 3 4 5 6 7 8 9 10 Israelachvili, J. N. (2011). Intermolecular and Surface Forces. Cambridge, Massachusetts: Academic Press.
- 1 2 3 4 5 6 7 8 9 10 11 12 Varughese, S.; Kiran, M. S. R. N.; Ramamurty, U.; Desiraju, G. R. (2013). "Nanoindentation in Crystal Engineering: Quantifying Mechanical Properties of Molecular Crystals". Angewandte Chemie International Edition. 52 (10): 2701–2712. doi:10.1002/anie.201205002. PMID 23315913.
- ↑ Field, J. E., ed. (1979). The Properties of Diamonds. New York, New York: Academic Press.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Haynes, W. M.; Lise, D. R.; Bruno, T. J., eds. (2016). CRC Handbook of Chemistry and Physics. Boca Raton, Florida: CRC Press.
- 1 2 O'Neil, M. J., ed. (2013). The Merck Index - An Encyclopedia of Chemicals, Drugs, and Biologicals. Cambridge, United Kingdom: Royal Society of Chemistry.
- ↑ Barret, C. S.; Meyer, L. (1965). Daunt, J. G. (ed.). Low Temperature Physics: The Crystal Structures of Argon and Its Alloys. New York, New York: Springer.
- ↑ Eisenberg, D.; Kauzmann, W. (2005). The Structures and Properties of Water. Oxford, UK: Oxford University Press.
- 1 2 Harvey, G. R. (1991). Polycyclic Aromatic Hydrocarbons: Chemistry and Carcinogenicity. Cambridge, UK: Cambridge University Press.
- 1 2 3 Jones, W., ed. (1997). Organic Molecular Solids: Properties and Applications. Boca Raton: CRC Press.
- 1 2 Desiraju, G. R. (2013). "Crystal Engineering: From Molecular to Crystal". Journal of the American Chemical Society. 135 (27): 9952–9967. doi:10.1021/ja403264c. PMID 23750552.
- ↑ Thakur, T. S.; Dubey, R.; Desiraju, G. R. (2015). "Crystal Structure and Prediction". Annual Review of Physical Chemistry. 1: 21–42. Bibcode:2015ARPC...66...21T. doi:10.1146/annurev-physchem-040214-121452. PMID 25422850.
- ↑ Davey, R. J.; Schroeder, S. L.; Horst, J. H. T. (2013). "Nucleation of Organic Crystals - A Molecular Perspective". Angewandte Chemie International Edition. 52 (8): 2166–2179. doi:10.1002/anie.201204824. PMID 23307268.
- ↑ Harris, Harris; Edward, M.; Blake, F. C. (1928). "The Atomic Arrangement of Orthorhombic Iodine". Journal of the American Chemical Society. 50 (6): 1583–1600. doi:10.1021/ja01393a009.
- ↑ Allan, D. R.; Clark, S. J.; Ibberson, R. M.; Parsons, S.; Pulham, C. R.; Sawyer, L. (1999). "The Influence of Pressure and Temperature on the Crystal Structure of Acetone". Chemical Communications (8): 751–752. doi:10.1039/A900558G. S2CID 54901610.
- ↑ Alt, H. C.; Kalus, J. (1982). "X-Ray Powder Diffraction Investigation of Naphthalene up to 0.5 GPa". Acta Crystallographica Section B. 38 (10): 2595–2600. doi:10.1107/s056774088200942x.
- 1 2 3 Williams, J. H. (1993). "The Molecular Electric QuadrupoleMoment and Solid-State Architecture". Accounts of Chemical Research. 26 (11): 593–598. doi:10.1021/ar00035a005.
- 1 2 Dawson, A.; Allan, D. R.; Parsons, Simon; Ruf, M. (2004). "Use of a CCD diffractometer in crystal structure determinations at high pressure". Journal of Applied Crystallography. 37 (3): 410–416. doi:10.1107/s0021889804007149.
- 1 2 3 Hassel, O.; Hvoslef, J. (1954). "The Structure of Bromine 1,4-Dioxanate". Acta Chemica Scandinavica. 8: 873. doi:10.3891/acta.chem.scand.08-0873.
- ↑ Metrangolo, P.; Meyer, F.; Pilati, Tullio; Resnati, G.; Terraneo, G. (2008). "Halogen Bonding in Supramolecular Chemistry". Angewandte Chemie International Edition. 47 (33): 6114–6127. doi:10.1002/anie.200800128. PMID 18651626.
- 1 2 3 4 5 6 7 8 Reddy, C. M.; Krishan, G. R.; Ghosh, S. (2010). "Mechanical properties of molecular crystals—applications to crystal engineering". CrystEngComm. 12 (8): 2296–2314. doi:10.1039/c003466e.
- 1 2 3 4 Kistenmacher, T. J.; Phillips, T. E.; Cowan, D. O. (1974). "The Crystal Structure of the 1:1 Radical Cation-Radical Anion Salt of 2,2'-bis-1,3-dithiole (TTF) and 7,7,8,8-tetracyanoquinodimethane (TCNQ)". Acta Crystallographica Section B. 30 (3): 763–768. doi:10.1107/s0567740874003669.
- ↑ Cohen, M. J.; Coleman, L. B.; Garito, A. F.; Heeger, A. J. (1974). "Electrical Conductivity of Tetrathiofulvalinium Tetracyanoquinodimethane (TTF) (TCNQ)". Physical Review B. 10 (4): 1298–1307. Bibcode:1974PhRvB..10.1298C. doi:10.1103/PhysRevB.10.1298.
- ↑ John Olmsted, Gregory M. Williams (1997). Chemistry: the molecular science. Jones & Bartlett Learning. p. 981. ISBN 978-0-8151-8450-8.
- ↑ Singhal Atul (2009). The Pearson Guide to Objective Chemistry for the AIEEE. Pearson Education India. p. 36. ISBN 978-81-317-1359-4.
- ↑ Gary Wulfsberg (1991). Principles of descriptive inorganic chemistry. University Science Books. p. 186. ISBN 978-0-935702-66-8.
- ↑ Simon, Arndt; Borrmann, Horst; Horakh, Jörg (1997). "On the Polymorphism of White Phosphorus". Chemische Berichte. 130 (9): 1235. doi:10.1002/cber.19971300911.
- ↑ AK Srivastava and PC Jain. Chemistry Vol (1 and 2). FK Publications. p. 548. ISBN 978-81-88597-83-3.
- ↑ Holleman, Arnold F; Wiberg, Egon; Wiberg, Nils (1985). "Arsen". Lehrbuch der Anorganischen Chemie (in German) (91–100 ed.). Walter de Gruyter. pp. 675–681. ISBN 978-3-11-007511-3.
- ↑ Masters, Anthony F. "Allotropes – Group 13, Group 14, Group 15, Group 16". Chemistry Explained. Retrieved 2009-01-06.
- ↑ James E. House (2008). Inorganic chemistry. Academic Press. p. 524. ISBN 978-0-12-356786-4.
- ↑ Darrell D. Ebbing, Steven D. Gammon (2007). General Chemistry. Cengage Learning. p. 446. ISBN 978-0-618-85748-7.
- ↑ James Wei (2007). Product engineering: molecular structure and properties. Oxford University Press. p. 137. ISBN 978-0-19-515917-2.
- ↑ Lide, D. R., ed. (2005). CRC Handbook of Chemistry and Physics (86th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- ↑ AK Srivastava and PC Jain. Chemistry Vol (1 and 2). FK Publications. p. 550. ISBN 978-81-88597-83-3.
- ↑ O. Gunnarsson (1997). "Superconductivity in Fullerides". Reviews of Modern Physics. 69 (2): 575. arXiv:cond-mat/9611150. Bibcode:1997RvMP...69..575G. doi:10.1103/RevModPhys.69.575. S2CID 18025631.