Unlike its lighter congeners, the halogen iodine forms a number of stable organic compounds, in which iodine exhibits higher formal oxidation states than -1 or coordination number exceeding 1. These are the hypervalent organoiodines, often called iodanes after the IUPAC rule used to name them.
These iodine compounds are hypervalent because the iodine atom formally contains in its valence shell more than the 8 electrons required for the octet rule. Hypervalent iodine oxyanions are known for oxidation states +1, +3, +5, and +7; organic analogues of these moieties are known for each oxidation state except +7.
In terms of chemical behavior, λ3‑ and λ5‑iodanes are generally oxidizing and/or electrophilic species. They have been widely applied towards those ends in organic synthesis.[1]
Nomenclature
Several different naming conventions are in use for the hypervalent organoiodines.
All begin with nonstandard formal charge assignments. In iodane chemistry, carbon is considered more electronegative than iodine, despite the Pauling electronegativities of those respective atoms.[2] Thus iodobenzene (C6H5I) is an iodine(I) compound, (dichloroiodo)benzene (C6H5ICl2) and iodosobenzene (C6H5IO) iodine(III) compounds, and iodoxybenzene (C6H5IO2) an iodine(V) compound.
With that convention in place, IUPAC names assume complete electron transfer. Thus when iodine is ligated to an organic residue and two Lewis acids, it is in the +3 oxidation state and the corresponding compound is a λ3‑iodane. A compound with iodine(V) would be a λ5‑iodane, and a hypothetical iodine(VII)‑containing compound would be a λ7‑iodane. Organyl-iodine ethers, a kind of λ3‑iodane, are sometimes called organic hypoiodites.
Alternatively, the hypervalent iodines can be classified using neutral electron counting. Iodine itself contains 7 valence electrons, and, in a monovalent iodane such as iodobenzene (C6H5I), the phenyl ligand donates one additional electron to give a completed octet. In a λ3‑iodane, each X-type ligand donates an additional electron, for 10 in total; the result is a decet structure. Similarly, many λ5‑iodanes are dodecet molecules, and hypothetical λ7‑iodanes are tetradecet molecules. As with other hypervalent compounds, N‑X‑L notation can be used to describe the formal electron count of iodanes, in which N stands for the number of electrons around the central atom X (in this case iodine), and L is the total number of ligand bonds with X. Thus, λ3‑iodanes can be described as 10‑I‑3 compounds, λ5‑iodanes as 12‑I‑5 compounds, and hypothetical λ7‑iodanes as 14‑I‑7 compounds.
Electron structure
As with other hypervalent compounds, iodanes bonding was formerly described using d-orbital participation. 3-center-4-electron bonding is now believed to be the primary bonding mode. This paradigm was developed by J.J. Musher in 1969.
One such bond exists in iodine(III) compounds, two such bonds reside in iodine(V) compounds and three such bonds would reside in the hypothetical iodine(VII) compounds.
Examples
Hypervalent organoiodine compounds are prepared by the oxidation of an organyl iodide.
In 1886, German chemist Conrad Willgerodt prepared the first hypervalent iodine compound, iodobenzene dichloride (PhICl2), by passing chlorine gas through iodobenzene in a cooled solution of chloroform:[3]
This preparation can be varied to produce iodobenzene pseudohalides. Cleaner preparations[4] begin with solutions of peracetic acid in glacial acetic acid, also due to Willgerodt:[5]
C6H5I + CH3C(O)OOH + CH3COOH → C6H5I(OC(O)CH3)2 + H2O
The iodobenzene diacetate product hydrolyzes to the polymeric iodosobenzene (PhIO), which is stable in cool alkaline solution.[6] In hot water (or, in Willgerodt's original preparation, steam distillation), iodosobenzene instead disproportionates to iodoxybenzene and iodobenzene:[7]
- 2 PhIO → PhIO2 + PhI
2-Iodobenzoic acid reacts with oxone[8] or a combination of potassium bromate and sulfuric acid to produce the insoluble λ5‑iodane 2-iodoxybenzoic (IBX) acid.[9] IBX acid is unstable and explosive, but acetylation tempers it to the stabler Dess-Martin periodinane.[10]
Aliphatic hypoiodites can be synthesized through a variant on the Williamson ether synthesis: an alkoxide reacts with iodine monochloride, releasing the alkyl hypoiodite and chloride.[11] Alternatively, the Meyer-Hartmann reaction applies: a silver alkoxide reacts with elemental iodine to give the hypoiodite and silver iodide. They are unstable to visible light, cleaving into alkoxyl and iodine radicals.[12]
The synthesis of organyl periodyl derivatives (λ7-iodanes) has been attempted since the early 20th century.[13] Efforts so far have met with failure, although aryl λ7‑chloranes are known. Organic diesters of iodine(VII) are presumed intermediates in the periodate cleavage of diols (Malaprade reaction), although no carbon-iodine(VII) bond is present in this process.
Diaryliodonium salts
Diaryliodonium salts are compounds of the type [Ar−I+−Ar]X−.[14] They are formally composed of a diaryliodonium cation[15] paired with a counteranion, but crystal structures show a long, weak, partially-covalent bond between the iodine and the counterion. Some authors have described this interaction as an example of halogen bonding,[16] but the interaction exists even with traditionally noncoordinating ions, such as perchlorate, triflate, or tetrafluoroborate.[17] As a result, other authors regard the diaryliodonia as λ3-iodanes.[18]
The salts are generally T-shaped, with the counteranion occupying an apical position.[18] The overall geometry at the iodine atom is pseudotrigonal bipyramidal. The placement of ligands exhibits apicophilicity: the phenyl group and chlorine group attain apical positions, while the other phenyl group and a lone pair of electrons hold equatorial ones.
Salts with a halide counterion are poorly soluble in many organic solvents, possibly because the halides bridge dimers. Solubility improves with triflate and tetrafluoroborate counterions.[17]
In general, the salts can be prepared from preformed hypervalent iodines such as iodic acid, iodosyl sulfate or iodosyl triflate. The first such compound was synthesised in 1894, via the silver hydroxide-catalyzed coupling of two aryl iodides (the Meyer–Hartmann reaction):[19][20][21]

Alternatively, the iodane may be formed in situ: an aryl iodide is oxidized to an aryliodine(III) compound (such as ArIO), followed by a ligand exchange. The latter can occur with organometallized arenes such as an arylstannane or -silane (a nucleophilic aromatic substitution reaction) or unfunctionalized arenes in the presence of a Brønsted or Lewis acid (an electrophilic aromatic substitution reaction).
Diaryliodonium salts react with nucleophiles at iodine, replacing one ligand to form the substituted arene ArNu and iodobenzene ArI. Diaryliodonium salts also react with metals M through ArMX intermediates in cross-coupling reactions.
Uses
Hypervalent iodine compounds are predominantly used as oxidizing reagents, although they are specialized and expensive. In some cases they replace more toxic oxidants.[23]
Iodobenzene diacetate (PhIAc2) and iodobenzene di(trifluoroacetate) are both strong oxidizing agents used in organic oxidations, as well as precursors for further organoiodine compounds. A hypervalent iodine (III) reagent was used as oxidant, together with ammonium acetate as nitrogen source, to provide 2-Furonitrile, a pharmaceutical intermediate and potential artificial sweetener.[24]
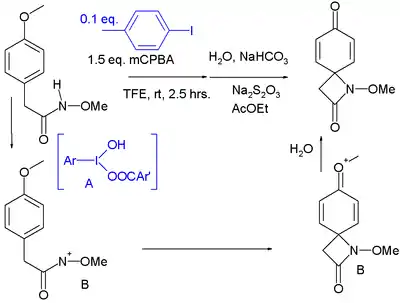
Current research focuses on the use of iodanes in carbon-carbon and carbon-heteroatom bond-forming reactions. In one study, an intramolecular C-N coupling of an alkoxyhydroxylamine to its anisole group is accomplished with a catalytic amount of aryliodide in trifluoroethanol.[25]
See also
References
- ↑ (Anastasios), Varvoglis, A. (1997). Hypervalent iodine in organic synthesis. London: Academic Press. ISBN 9780127149752. OCLC 162128812.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ↑ However, iodanes usually feature bonds to carbon in its sp2- or sp-hybridized state. The hybridization-specific electronegativities of sp2 and sp carbon are estimated to be 3.0 and 3.3, respectively (Anslyn and Dougherty, Modern Physical Organic Chemistry, University Science Books, 2004).
- ↑ C. Willgerodt, Tageblatt der 58. Vers. deutscher Naturforscher u. Aertzte, Strassburg 1885.
- ↑ J. G. Sharefkin and H. Saltzman. "Benzene, iodoso-, diacetate". Organic Syntheses.; Collective Volume, vol. 5, p. 660
- ↑ Willgerodt, C. (1892). "Zur Kenntniss aromatischer Jodidchloride, des Jodoso- und Jodobenzols". Chem. Ber. (in German). 25 (2): 3494–3502. doi:10.1002/cber.189202502221.
- ↑ H. Saltzman and J. G. Sharefkin. "Benzene, iodoso-". Organic Syntheses.; Collective Volume, vol. 5, p. 658
- ↑ J. G. Sharefkin and H. Saltzman. "Benzene, iodoxy-". Organic Syntheses.; Collective Volume, vol. 5, p. 665
- ↑ Frigerio, M.; Santagostino, M.; Sputore, S. (1999). "A User-Friendly Entry to 2-Iodoxybenzoic Acid (IBX)". J. Org. Chem. 64 (12): 4537–4538. doi:10.1021/jo9824596.
- ↑ Robert K. Boeckman, Jr., Pengcheng Shao, and Joseph J. Mullins. "1,2-Benziodoxol-3(1H)-one, 1,1,1-tris(acetyloxy)-1,1-dihydro-". Organic Syntheses.
{{cite journal}}: CS1 maint: multiple names: authors list (link); Collective Volume, vol. 10, p. 696 - ↑ Dess, D. B.; Martin, J. C. (1983). "Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones". J. Org. Chem. 48 (22): 4155–4156. doi:10.1021/jo00170a070.
- ↑ Glover, Stephen A.; Goosen, André (January 1980). "Synthesis of β=iodo--butyl and methyl ethers from the reaction of alkenes with -butyl and methyl hypoiodites". Tetrahedron Letters. 21 (20): 2005–2008. doi:10.1016/S0040-4039(00)93669-4.
- ↑ Beebe, Thomas R.; Barnes, Beverly A.; Bender, Keith A.; Halbert, Allan D.; Miller, Robert D.; Ramsay, Martin L.; Ridenour, Michael W. (June 1975). "Oxidation of alcohols with acetyl hypoiodite". The Journal of Organic Chemistry. 40 (13): 1992–1994. doi:10.1021/jo00901a028. ISSN 0022-3263.
- ↑ Luliński, Piotr; Sosnowski, Maciej; Skulski, Lech; Luliński, Piotr; Sosnowski, Maciej; Skulski, Lech (2005-05-13). "A Novel Aromatic Iodination Method, with Sodium Periodate Used as the Only Iodinating Reagent". Molecules. 10 (3): 516–520. doi:10.3390/10030516. PMC 6147649. PMID 18007324.
- ↑ Merritt, Eleanor A.; Olofsson, Berit (2009). "Diaryliodonium Salts: A Journey from Obscurity to Fame". Angew. Chem. Int. Ed. 48 (48): 9052–9070. doi:10.1002/anie.200904689. PMID 19876992.
- ↑ Note that in the diaryliodonium salt description, the compound is not hypervalent, and the bonding number is the standard one for iodine (λ1). It is a 8-I-2 species. In the other common description of these compounds as covalent iodanes, they are formally 10-I-3 and λ3.
- ↑ Resnati, G.; Ursini, M.; Pilati, T.; Politzer, P.; Murray, J. S.; Cavallo, G. (2017-07-01). "Halogen bonding in hypervalent iodine and bromine derivatives: halonium salts". IUCrJ. 4 (4): 411–419. doi:10.1107/S2052252517004262. ISSN 2052-2525. PMC 5571804. PMID 28875028.
- 1 2 Neckers, Douglas C.; Pinkerton, A. Alan; Gu, Haiyan; Kaafarani, Bilal R. (2002-05-28). "The crystal and molecular structures of 1-naphthylphenyliodonium tetrafluoroborate and 1-naphthylphenyliodonium tetrakis(pentafluorophenyl)gallate". Journal of the Chemical Society, Dalton Transactions (11): 2318–2321. doi:10.1039/B202805K. ISSN 1364-5447.
- 1 2 "Iodonium salts in organic synthesis". www.arkat-usa.org. Retrieved 2018-12-30.
- ↑ Hartmann, Christoph; Meyer, Victor (1894). "Ueber die Jodoniumbasen". Berichte der Deutschen Chemischen Gesellschaft (in German). 27 (1): 502–509. doi:10.1002/cber.18940270199.
- ↑ Bothner-By, Aksel A.; Vaughan, C. Wheaton Jr. (1952). "The Gross Mechanism of the Victor Meyer and Hartmann Reaction". J. Am. Chem. Soc. 74 (17): 4400–4401. doi:10.1021/ja01137a048.
- ↑ Wang, Zerong (2010). "Meyer–Hartmann Reaction". Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons, Inc. pp. 1910–1912. doi:10.1002/9780470638859.conrr429. ISBN 9780470638859.
- ↑ Dohi, T.; Maruyama, A.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. (2007). "First hypervalent iodine(III)-catalyzed C-N bond forming reaction: catalytic spirocyclization of amides to N-fused spirolactams". Chemical Communications. 44 (12): 1224–1226. doi:10.1039/b616510a. PMID 17356763.
- ↑ Hypervalent iodine(V) reagents in organic synthesis Uladzimir Ladziata and Viktor V. Zhdankin Arkivoc 05-1784CR pp 26-58 2006 Article
- ↑ Chenjie Zhu; Sun, Chengguo; Wei, Yunyang (2010). "Direct oxidative conversion of alcohols, aldehydes and amines into nitriles using hypervalent iodine(III) reagent". Synthesis. 2010 (24): 4235–4241. doi:10.1055/s-0030-1258281.
- ↑ Dohi, T.; Maruyama, A.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. (2007). "First hypervalent iodine(III)-catalyzed C-N bond forming reaction: catalytic spirocyclization of amides to N-fused spirolactams". Chemical Communications. 44 (12): 1224–1226. doi:10.1039/b616510a. PMID 17356763.