Hydrogen–deuterium exchange (also called H–D or H/D exchange) is a chemical reaction in which a covalently bonded hydrogen atom is replaced by a deuterium atom, or vice versa. It can be applied most easily to exchangeable protons and deuterons, where such a transformation occurs in the presence of a suitable deuterium source, without any catalyst. The use of acid, base or metal catalysts, coupled with conditions of increased temperature and pressure, can facilitate the exchange of non-exchangeable hydrogen atoms, so long as the substrate is robust to the conditions and reagents employed. This often results in perdeuteration: hydrogen-deuterium exchange of all non-exchangeable hydrogen atoms in a molecule.
An example of exchangeable protons which are commonly examined in this way are the protons of the amides in the backbone of a protein.[1][2][3] The method gives information about the solvent accessibility of various parts of the molecule, and thus the tertiary structure of the protein. The theoretical framework for understanding hydrogen exchange in proteins was first described by Kaj Ulrik Linderstrøm-Lang and he was the first to apply H/D exchange to study proteins.[4]
Exchange reaction
In protic solution exchangeable protons such as those in hydroxyl or amine group exchange protons with the solvent. If D2O is solvent, deuterons will be incorporated at these positions. The exchange reaction can be followed using a variety of methods (see Detection). Since this exchange is an equilibrium reaction, the molar amount of deuterium should be high compared to the exchangeable protons of the substrate. For instance, deuterium is added to a protein in H2O by diluting the H2O solution with D2O (e.g. tenfold). Usually exchange is performed at physiological pH (7.0–8.0) where proteins are in their most native ensemble of conformational states.[5][6]
The H/D exchange reaction can also be catalysed, by acid, base or metal catalysts such as platinum. For the backbone amide hydrogen atoms of proteins, the minimum exchange rate occurs at approximately pH 2.6, on average. By performing the exchange at neutral pH and then rapidly changing the pH, the exchange rates of the backbone amide hydrogens can be dramatically slowed, or quenched. The pH at which the reaction is quenched depends on the analysis method. For detection by NMR, the pH may be moved to around 4.0–4.5. For detection by mass spectrometry, the pH is dropped to the minimum of the exchange curve, pH 2.6. In the most basic experiment, the reaction is allowed to take place for a set time before it is quenched.
The deuteration pattern of a molecule that has undergone H/D exchange can be maintained in aprotic environments. However, some methods of deuteration analysis for molecules such as proteins, are performed in aqueous solution, which means that exchange will continue at a slow rate even after the reaction is quenched. Undesired deuterium-hydrogen exchange is referred to as back-exchange and various methods have been devised to correct for this.
Detection
H–D exchange was measured originally by the father of hydrogen exchange Kaj Ulrik Linderstrøm-Lang using density gradient tubes. In modern times, H–D exchange has primarily been monitored by the methods: NMR spectroscopy, mass spectrometry and neutron crystallography. Each of these methods have their advantages and drawbacks.
NMR spectroscopy
Hydrogen and deuterium nuclei are grossly different in their magnetic properties. Thus it is possible to distinguish between them by NMR spectroscopy. Deuterons will not be observed in a 1H NMR spectrum and conversely, protons will not be observed in a 2H NMR spectrum. Where small signals are observed in a 1H NMR spectrum of a highly deuterated sample, these are referred to as residual signals. They can be used to calculate the level of deuteration in a molecule. Analogous signals are not observed in 2H NMR spectra because of the low sensitivity of this technique compared to the 1H analysis. Deuterons typically exhibit very similar chemical shifts to their analogous protons. Analysis via 13C NMR spectroscopy is also possible: the different spin values of hydrogen (1/2) and deuterium (1) gives rise to different splitting multiplicities. NMR spectroscopy can be used to determine site-specific deuteration of molecules.
Another method uses HSQC spectra. Typically HSQC spectra are recorded at a series of timepoints while the hydrogen is exchanging with the deuterium. Since the HSQC experiment is specific for hydrogen, the signal will decay exponentially as the hydrogen exchanges. It is then possible to fit an exponential function to the data, and obtain the exchange constant. This method gives residue-specific information for all the residues in the protein simultaneously[7][8] The major drawback is that it requires a prior assignment of the spectrum for the protein in question. This can be very labor-intensive, and usually limits the method to proteins smaller than 25 kDa. Because it takes minutes to hours to record a HSQC spectrum, amides that exchange quickly must be measured using other pulse sequences.
Mass spectrometry
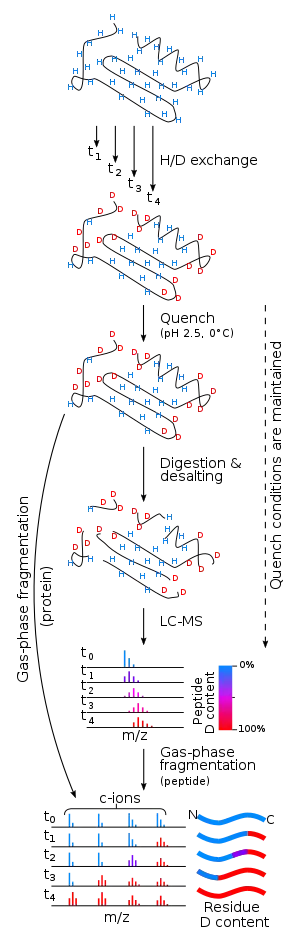
Hydrogen–deuterium exchange mass spectrometry (HX-MS or HDX-MS) can determine the overall deuterium content of molecules which have undergone H/D exchange. Because of the sample preparation required, it is typically considered to provide an accurate measurement of non-exchangeable hydrogen atoms only. It can also involve H/D exchange in the gas phase[9] or solution phase exchange prior to ionization.[3] It has several advantages in NMR spectroscopy with respect to analysis of H–D exchange reactions: much less material is needed, the concentration of sample can be very low (as low as 0.1 uM), the size limit is much greater, and data can usually be collected and interpreted much more quickly.[10]
The deuterium nucleus is twice as heavy as the hydrogen nucleus because it contains a neutron as well as a proton. Thus a molecule that contains some deuterium will be heavier than one that contains all hydrogen. As a protein is increasingly deuterated, the molecular mass increases correspondingly. Detecting the change in the mass of a protein upon deuteration was made possible by modern protein mass spectrometry, first reported in 1991 by Katta and Chait.[11]
Determining site specific deuteration via mass spectrometry is more complicated than using NMR spectroscopy. For example, the location and relative amount of deuterium exchange along the peptide backbone can be determined roughly by subjecting the protein to proteolysis after the exchange reaction has been quenched. Individual peptides are then analyzed for overall deuteration of each peptide fragment. Using this technique the resolution of deuterium exchange is determined by the size of the peptides produced during digestion.[12] Pepsin, an acid protease, is commonly used for proteolysis, as the quench pH must be maintained during the proteolytic reaction. To minimize the back-exchange, proteolysis and subsequent mass spectrometry analysis must be done as quickly as possible. HPLC separation of the peptic digest is often carried out at low temperature just prior to electrospray mass spectrometry to minimize back-exchange. More recently, UPLC has been used due to its superior separation capabilities.[13]
It was proposed in 1999 that it might be possible to achieve single-residue resolution by using collision-induced dissociation (CID) fragmentation of deuterated peptides in conjunction with tandem mass spectrometry. It was soon discovered that CID causes "scrambling" of the deuterium position within the peptides.[14][15] However, fragmentation produced by MALDI in-source decay (ISD), electron capture dissociation (ECD), and electron transfer dissociation (ETD) proceed with little or no scrambling under the correct experimental conditions.[16][17][18] Scrambling of the isotopic labeling is caused by collisional heating prior to dissociation of the ion and while CID do cause scrambling, collisional heating can also occur during ionization and ion transport.[19] However, by careful optimization of instrument parameters which cause ion heating, hydrogen scrambling can be minimized to a degree which preserves the solution phase isotopic labeling until fragmentation can be performed using a technique where scrambling does not occur.[17][18][20][21] More recently, ultraviolet photodissociation (UVPD) has also been investigated as a possible fragmentation technique to localize deuterium within peptides and proteins.[22][23] In this regard, the conclusions have been mixed, while it is possible to obtain UVPD fragments which has not undergone scrambling under certain conditions, others have shown that scrambling can occur for both peptides and proteins during the UVPD fragmentation step itself.[22][23] The theory consolidating these apparent contradictions has to do with the dual fragmentation pathway that may arise from UV irradiation of peptides and proteins, i.e. direct and statistical dissociation.[24] That is, if experimental conditions favor direct dissociation and the precursor ion is kept at low internal energies before and during fragmentation the deuterium level of the resulting fragments will correspond to the non-scrambled precursor.[22] However, experimental conditions may favor statistical dissociation during UV irradiation, especially at long irradiation times and low gas pressure, leading to internal conversion of the electronic excitation energy contributed by the UV photons.[23] The result is vibrational excitation of the irradiated molecule which in turn undergo scrambling.
Neutron crystallography
Hydrogen–deuterium exchange of fast-exchanging species (e.g. hydroxyl groups) can be measured at atomic resolution quantitatively by neutron crystallography, and in real time if exchange is conducted during the diffraction experiment.
High intensity neutron beams are generally generated by spallation at linac particle accelerators such as the Spallation Neutron Source. Neutrons diffract crystals similarly to X-rays and can be used for structural determination. Hydrogen atoms, with between one and zero electrons in a biological setting, diffract X-rays poorly and are effectively invisible under normal experimental conditions. Neutrons scatter from atomic nuclei, and are therefore capable of detecting hydrogen and deuterium atoms.
Hydrogen atoms are routinely replaced with deuterium, which introduce a strong and positive scattering factor. It is often sufficient to replace only the solvent and labile hydrogen atoms in a protein crystal by vapor diffusion. In such a structure the occupancy of an exchangeable deuterium atom in a crystal will refine from 0-100%, directly quantifying the amount of exchange.
Applications
Neutron scattering
Perdeuteration of one component of a multi-component system can provide contrast for neutron scattering experiments, where the contrast obtained by using deuterated solvents is insufficient.
Protein structure
It is not possible to determine the structure of a protein with H/D exchange other than neutron crystallography nor is it possible to define secondary structural elements. The reasons for this are related to the way in which protein structure slows exchange. Exchange rates are a function of two parameters: solvent accessibility and hydrogen bonding. Thus an amide which is part of an intramolecular hydrogen bond will exchange slowly if at all, while an amide on the surface of protein hydrogen bonded to water will exchange rapidly. Amides buried from the solvent but not hydrogen bonded may also have very slow exchange rates. Because both solvent accessibility and hydrogen bonding contribute to the rate of exchange, it becomes difficult to attribute a given exchange rate to a structural element without crystallography or NMR structural data.
H–D exchange has been used to characterize the folding pathway of proteins, by refolding the protein under exchange conditions. In a forward exchange experiment (H to D), a pulse of deuterium is added after various amounts of refolding time. The parts of the structure that form rapidly will be protected and thus not exchanged, whereas areas that fold late in the pathway will be exposed to the exchange for longer periods of time. Thus H/D exchange can be used to determine the sequence of various folding events. Factors determining the time resolution of this approach are the efficiency of mixing and how quickly the quench can be performed after the labeling.
H–D exchange has been used to characterize protein structures[25] and protein–protein interactions.[26] The exchange reaction needs to be carried out with the isolated proteins and with the complex. The exchanging regions are then compared. If a region is buried by the binding, the amides in this region may be protected in the complex and exchange slowly. However, one must bear in mind that H–D exchange cannot be used to locate binding interfaces for all protein-protein interactions. Some protein-protein interactions are driven by electrostatic forces of side chains and are unlikely to change the exchange rate of backbone amide hydrogens, particularly if the amide hydrogens are located in stable structural elements such as alpha helices.
Lastly, H–D exchange can be used to monitor conformational changes in proteins as they relate to protein function. If conformation is altered as result of post-translational modification, enzyme activation, drug binding or other functional events, there will likely be a change to H/D exchange that can be detected.[27]
HDXsite online webserver
HDXsite is an online websever which includes some applications such as HDX modeller increasing the resolution of experimental HDX data and modeling protection factors for individual residues.[28] [29]
References
- ↑ Englander SW, Mayne L (1992-06-01). "Protein folding studied using hydrogen-exchange labeling and two-dimensional NMR". Annual Review of Biophysics and Biomolecular Structure. 21 (1): 243–65. doi:10.1146/annurev.bb.21.060192.001331. PMID 1525469.
- ↑ Englander SW, Sosnick TR, Englander JJ, Mayne L (February 1996). "Mechanisms and uses of hydrogen exchange". Current Opinion in Structural Biology. 6 (1): 18–23. doi:10.1016/s0959-440x(96)80090-x. PMC 3412065. PMID 8696968.
- 1 2 Konermann L, Pan J, Liu YH (March 2011). "Hydrogen exchange mass spectrometry for studying protein structure and dynamics". Chemical Society Reviews. 40 (3): 1224–34. doi:10.1039/C0CS00113A. PMID 21173980.
- ↑ Englander SW, Mayne L, Bai Y, Sosnick TR (May 1997). "Hydrogen exchange: the modern legacy of Linderstrøm-Lang". Protein Science. 6 (5): 1101–9. doi:10.1002/pro.5560060517. PMC 2143687. PMID 9144782.
- ↑ Englander SW, Kallenbach NR (November 1983). "Hydrogen exchange and structural dynamics of proteins and nucleic acids". Quarterly Reviews of Biophysics. 16 (4): 521–655. doi:10.1017/S0033583500005217. PMID 6204354. S2CID 37407781.
- ↑ Johnson RS, Walsh KA (December 1994). "Mass Spectrometric Measurement of Protein Amide Hydrogen Exchange of Apo- and Holo-Myoglobin". Protein Science. 3 (12): 2411–2418. doi:10.1002/pro.5560031224. PMC 2142783. PMID 7756994.
- ↑ Demura M (2006). "NMR Insight of Structural Stability and Folding of Calcium-Binding Lysozyme". In Webb GA (ed.). Modern Magnetic Resonance. Dordrecht: Springer. pp. 497–501. doi:10.1007/1-4020-3910-7_62. ISBN 978-1-4020-3894-5.
- ↑ Chandak MS, Nakamura T, Makabe K, Takenaka T, Mukaiyama A, Chaudhuri TK, et al. (July 2013). "The H/D-exchange kinetics of the Escherichia coli co-chaperonin GroES studied by 2D NMR and DMSO-quenched exchange methods". Journal of Molecular Biology. 425 (14): 2541–60. doi:10.1016/j.jmb.2013.04.008. PMID 23583779.
- ↑ Stewart JH, Shapiro RH, DePuy CH, Bierbaum VH (1977). "Hydrogen-deuterium exchange reactions of carbanions with water-d2 in the gas phase". Journal of the American Chemical Society. 99 (23): 7650–3. doi:10.1021/ja00465a037.
- ↑ Wales TE, Engen JR (2006). "Hydrogen exchange mass spectrometry for the analysis of protein dynamics". Mass Spectrometry Reviews. 25 (1): 158–70. Bibcode:2006MSRv...25..158W. doi:10.1002/mas.20064. PMID 16208684.
- ↑ Katta V, Chait BT (April 1991). "Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry". Rapid Communications in Mass Spectrometry. 5 (4): 214–7. Bibcode:1991RCMS....5..214K. doi:10.1002/rcm.1290050415. PMID 1666528.
- ↑ Zhang Z, Smith DL (April 1993). "Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation". Protein Science. 2 (4): 522–31. doi:10.1002/pro.5560020404. PMC 2142359. PMID 8390883.
- ↑ Wales TE, Fadgen KE, Gerhardt GC, Engen JR (September 2008). "High-speed and high-resolution UPLC separation at zero degrees Celsius". Analytical Chemistry. 80 (17): 6815–20. doi:10.1021/ac8008862. PMC 2562353. PMID 18672890.
- ↑ Jørgensen TJ, Gårdsvoll H, Ploug M, Roepstorff P (March 2005). "Intramolecular migration of amide hydrogens in protonated peptides upon collisional activation". Journal of the American Chemical Society. 127 (8): 2785–93. doi:10.1021/ja043789c. PMID 15725037.
- ↑ Jørgensen TJ, Bache N, Roepstorff P, Gårdsvoll H, Ploug M (December 2005). "Collisional activation by MALDI tandem time-of-flight mass spectrometry induces intramolecular migration of amide hydrogens in protonated peptides". Molecular & Cellular Proteomics. 4 (12): 1910–9. doi:10.1074/mcp.M500163-MCP200. PMID 16127176.
- ↑ Bache N, Rand KD, Roepstorff P, Jørgensen TJ (August 2008). "Gas-phase fragmentation of peptides by MALDI in-source decay with limited amide hydrogen (1H/2H) scrambling". Analytical Chemistry. 80 (16): 6431–5. doi:10.1021/ac800902a. PMID 18642878.
- 1 2 Rand KD, Adams CM, Zubarev RA, Jørgensen TJ (January 2008). "Electron capture dissociation proceeds with a low degree of intramolecular migration of peptide amide hydrogens". Journal of the American Chemical Society. 130 (4): 1341–9. doi:10.1021/ja076448i. PMID 18171065.
- 1 2 Zehl M, Rand KD, Jensen ON, Jørgensen TJ (December 2008). "Electron transfer dissociation facilitates the measurement of deuterium incorporation into selectively labeled peptides with single residue resolution". Journal of the American Chemical Society. 130 (51): 17453–9. doi:10.1021/ja805573h. PMID 19035774.
- ↑ Rand KD, Zehl M, Jørgensen TJ (October 2014). "Measuring the hydrogen/deuterium exchange of proteins at high spatial resolution by mass spectrometry: overcoming gas-phase hydrogen/deuterium scrambling". Accounts of Chemical Research. 47 (10): 3018–27. doi:10.1021/ar500194w. PMID 25171396.
- ↑ Wollenberg DT, Pengelley S, Mouritsen JC, Suckau D, Jørgensen CI, Jørgensen TJ (June 2020). "Avoiding H/D Scrambling with Minimal Ion Transmission Loss for HDX-MS/MS-ETD Analysis on a High-Resolution Q-TOF Mass Spectrometer" (PDF). Analytical Chemistry. 92 (11): 7453–7461. doi:10.1021/acs.analchem.9b05208. PMID 32427467. S2CID 218759759.
- ↑ Rand KD, Pringle SD, Morris M, Engen JR, Brown JM (October 2011). "ETD in a traveling wave ion guide at tuned Z-spray ion source conditions allows for site-specific hydrogen/deuterium exchange measurements". Journal of the American Society for Mass Spectrometry. 22 (10): 1784–93. Bibcode:2011JASMS..22.1784R. doi:10.1007/s13361-011-0196-7. PMC 3438897. PMID 21952892.
- 1 2 3 Mistarz UH, Bellina B, Jensen PF, Brown JM, Barran PE, Rand KD (January 2018). "UV Photodissociation Mass Spectrometry Accurately Localize Sites of Backbone Deuteration in Peptides". Analytical Chemistry. 90 (2): 1077–1080. doi:10.1021/acs.analchem.7b04683. PMID 29266933.
- 1 2 3 Modzel M, Wollenberg DT, Trelle MB, Larsen MR, Jørgensen TJ (December 2020). "Ultraviolet Photodissociation of Protonated Peptides and Proteins Can Proceed with H/D Scrambling" (PDF). Analytical Chemistry. 93 (2): 691–696. doi:10.1021/acs.analchem.0c02957. PMID 33295747. S2CID 228078945.
- ↑ R Julian R (September 2017). "The Mechanism Behind Top-Down UVPD Experiments: Making Sense of Apparent Contradictions". Journal of the American Society for Mass Spectrometry. 28 (9): 1823–1826. Bibcode:2017JASMS..28.1823J. doi:10.1007/s13361-017-1721-0. PMC 5711567. PMID 28702929.
- ↑ White MR, Khan MM, Deredge D, Ross CR, Quintyn R, Zucconi BE, Wysocki VH, Wintrode PL, Wilson GM, Garcin ED (January 2015). "A dimer interface mutation in glyceraldehyde-3-phosphate dehydrogenase regulates its binding to AU-rich RNA". The Journal of Biological Chemistry. 290 (3): 1770–85. doi:10.1074/jbc.M114.618165. PMC 4340419. PMID 25451934.
- ↑ Mandell JG, Baerga-Ortiz A, Falick AM, Komives EA (2005). "Measurement of solvent accessibility at protein-protein interfaces". Protein-Ligand Interactions. Methods in Molecular Biology. Vol. 305. pp. 65–80. doi:10.1385/1-59259-912-5:065. ISBN 1-59259-912-5. PMID 15939994.
- ↑ Guttman M, Cupo A, Julien JP, Sanders RW, Wilson IA, Moore JP, Lee KK (February 2015). "Antibody potency relates to the ability to recognize the closed, pre-fusion form of HIV Env". Nature Communications. 6: 6144. Bibcode:2015NatCo...6.6144G. doi:10.1038/ncomms7144. PMC 4338595. PMID 25652336.
- ↑ Salmas RE, Borysik AJ (February 2021). "HDXmodeller: an online webserver for high-resolution HDX-MS with auto-validation". Communications Biology. 4 (1): 199. doi:10.1038/s42003-021-01709-x. PMC 7884430. PMID 33589746.
- ↑ Salmas RE, Borysik AJ (May 2021). "Characterization and Management of Noise in HDX-MS Data Modeling". Analytical Chemistry. 93 (19): 7323–7331. doi:10.1021/acs.analchem.1c00894. PMID 33961396. S2CID 233998906.
Further reading
- David Weis, Ed. (2016). Hydrogen Exchange Mass Spectrometry of Proteins: Fundamentals, Methods, and Applications. New York: Wiley. ISBN 9781118616499.
- Masson GR, Burke JE, Ahn NG, Anand GS, Borchers C, Brier S, et al. (July 2019). "Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments". Nature Methods. 16 (7): 595–602. doi:10.1038/s41592-019-0459-y. PMC 6614034. PMID 31249422.