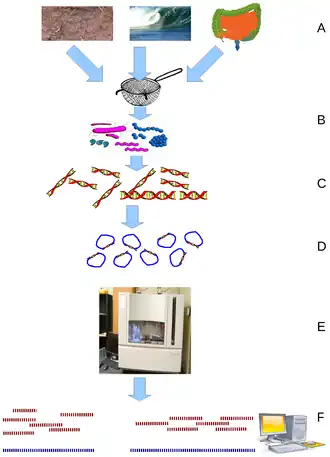
(B) filtering particles, typically by size
(C) Lysis and DNA extraction
(D) cloning and library construction
(E) sequencing the clones
(F) sequence assembly into contigs and scaffolds
Viral metagenomics uses metagenomic technologies to detect viral genomic material from diverse environmental and clinical samples.[1][2] Viruses are the most abundant biological entity and are extremely diverse; however, only a small fraction of viruses have been sequenced and only an even smaller fraction have been isolated and cultured.[1][3] Sequencing viruses can be challenging because viruses lack a universally conserved marker gene so gene-based approaches are limited.[3][4] Metagenomics can be used to study and analyze unculturable viruses and has been an important tool in understanding viral diversity and abundance and in the discovery of novel viruses.[1][5][6] For example, metagenomics methods have been used to describe viruses associated with cancerous tumors and in terrestrial ecosystems.[7]
History
The traditional methods for discovering, characterizing, and assigning viral taxonomy to viruses were based on isolating the virus particle or its nucleic acid from samples.[8] The virus morphology could be visualized using electron microscopy but only if the virus could be isolated in high enough titer to be detected. The virus could be cultured in eukaryotic cell lines or bacteria but only if the appropriate host cell type was known and the nucleic acid of the virus would be detected using PCR but only if a consensus primer was known.[8]
Metagenomics requires no prior knowledge of the viral genome as it does not require a universal marker gene, a primer or probe design.[9] Because this method uses prediction tools to detect viral content of a sample, it can be used to identify new virus species or divergent members of known species.
The earliest metagenomic studies of viruses were carried out on ocean samples in 2002. The sequences that were matched to referenced sequences were predominantly double-stranded DNA bacteriophages and double-stranded algal viruses.[10]
In 2016 the International Committee on Taxonomy of Viruses (ICTV) officially recognized that viral genomes assembled from metagenomic data can be classified using the same procedures for viruses isolated via classical virology approaches.[11]
Challenges
Viral dark matter
In the 2002 metagenomics study the researchers found that 65% of the sequences of DNA and RNA viruses had no matches in the reference databases.[10]This phenomenon of unmatched viral sequences in sequence reference databases is prevalent in viral metagenomics studies and is referred to as “viral dark matter".[3][8] It is predominantly caused by the lack of complete viral genome sequences of diverse samples in reference databases and the rapid rate of viral evolution.[3][8]
Virus nucliec acid type diversity
Adding to these challenges, there are seven classes of viruses based on the Baltimore classification system which groups viruses based on their genomic structure and their manner of transcription: there are double-stranded DNA viruses, single-stranded DNA viruses, double-stranded RNA viruses, and single-stranded RNA virus.[12] Single-stranded RNA can be positive or negative sense. These different nucleic acids types need different sequencing approaches and there is no universal gene marker that is conserved for all virus types.[3][4] Gene-based approaches can only target specific groups of viruses (such as RNA viruses that share a conserved RNA polymerase sequence).[3][4]
DNA virus bias
There is still a bias towards DNA viruses in reference databases. Common reasons for this bias is because RNA viruses mutate more rapidly than DNA viruses, DNA is easier to handle from samples while RNA is unstable, and more steps are needed for RNA metagenomics analysis (reverse transcription).[4][8]
Sequence contamination
Sequences can be contaminated with the host organism's' sequences which is particularly troublesome if the host organism of the virus is unknown.[4] There could also be contamination from nucleic acid extraction and PCR.[4]
Methods
Untargeted metagenomics
Metagenomic analysis uses whole genome shotgun sequencing to characterize microbial diversity in clinical and environmental samples. Total DNA and/or RNA are extracted from the samples and are prepared on a DNA or RNA library for sequencing.[9] These methods have been used to sequence the whole genome of Epstein–Barr virus (EBV) and HCV, however, contaminating host nucleic acids can affect the sensitivity to the target viral genome with the proportion of reads related to the target sequence often being low.[13][14]
The IMG/VR system and the IMG/VR v.2.0 are the largest interactive public virus databases with over 760,000 metagenomic viral sequences and isolate viruses and serves as a starting point for the sequence analysis of viral fragments derived from metagenomic samples.[15][16]
Targeted metagenomics: amplicon sequencing
While untargeted metagenomics and metatranscriptomics does not need a genetic marker, amplicon sequencing does. It uses a gene that is highly conserved as a genetic marker, but because of the varied nucleic acid types, the marker used has to be for specific groups of viruses.[3][4] This is done via PCR amplification of primers that are complementary to a known, highly conserved nucleotide sequence.[9] PCR is then followed by whole genome sequencing methods and has been used to track the Ebola virus,[17] Zika Virus,[18] and COVID-19[19] epidemics. PCR amplicon sequencing is more successful for whole genome sequencing of samples with low concentrations. However, with larger viral genomes and the heterogeneity of RNA viruses multiple overlapping primers may be required to cover the amplification of all genotypes. PCR amplicon sequencing requires knowledge of the viral genome prior to sequencing, appropriate primers, and is highly dependent on viral titers, however, PCR amplicon sequencing is a cheaper evaluation method than metagenomic sequencing when studying known viruses with relatively small genomes.[9]
Targeted metagenomics: enrichment with probes
Target enrichment is a culture independent method that sequences viral genomes directly from a sample using small RNA or DNA probes complementary to the pathogens reference sequence. The probes, which can be bound to a solid phase and capture and pull down complementary DNA sequences in the sample.[9] The presence of overlapping probes increases the tolerance for primer mismatches but their design requires high cost and time so a rapid response is limited. DNA capture is followed by brief PCR cycling and shotgun sequencing. Success of this method is dependent available reference sequences to create the probes and is not suitable for characterization of novel viruses.[9] This method has been used to characterize large and small viruses such as HCV,[14] HSV-1,[20] and HCMV.[21]
Limitations
Viral metagenomics methods can produce erroneous chimerical sequences.[22][23] These can include in vitro artifacts from amplification and in silico artifacts from assembly.[23] Chimeras can form between unrelated viruses, as well as between viral and eukaryotic sequences.[23] The likelihood of errors is partially mitigated by greater sequencing depth, but chimeras can still form in areas of high coverage if the reads are highly fragmented.[22]
Applications
Agriculture
Plant viruses pose a global threat to crop production but through metagenomic sequencing and viral database creation, modified plant viruses can be used to aid in plant immunity as well as alter physical appearance.[24] Data obtained on plant virus genomes from metagenomic sequencing can be used to create clone viruses to inoculate the plant with to study viral components and biological characterization of viral agents with increased reproducibility. Engineered mutant virus strains have been used to alter the coloration and size of various ornamental plants and promote the health of crops.[25]
Ecology
Viral metagenomics contributes to viral classification without the need of culture based methodologies and has provided vast insights on viral diversity in any system. Metagenomics can be used to study viruses effects on a given ecosystem and how they effect the microbiome as well as monitoring viruses in an ecosystem for possible spillover into human populations.[1] Within the ecosystems, viruses can be studied to determine how they compete with each other as well as viral effects on functions of the host. Viral metagenomics has been used to study unculturable viral communities in marine and soil ecosystems.[7][26]
Infectious disease research
Viral metagenomics is readily used to discover novel viruses, with a major focus on those zoonotic or pathogenic to humans. Viral databases obtained from metagenomics provides quick response methods to determine viral infections as well as determine drug resistant variants in clinical samples.[9] The contributions of viral metagenomics to viral classification have aided pandemic surveillance efforts as well as made infectious disease surveillance and testing more affordable.[27] Since the majority of human pandemics are zoonotic in origin, metagenomic surveillance can provide faster identification of novel viruses and their reservoirs.[28]
One such surveillance program is the Global Virome Project (GVP) an international collaborative research initiative based at the One Health Institute at the University of California, Davis.[29][30] The GVP aims to boost infectious disease surveillance around the globe by using low cost sequencing methods in high risk countries to prevent disease outbreaks and to prevent future virus outbreaks.[27][31]
Medicine
Viral metagenomics has been used to test for virus related cancers and difficult to diagnose cases in clinical diagnostics.[31] This method is most often used when conventional and advanced molecular testing cannot find a causative agent for disease. Metagenomic sequencing can also be used to detect pathogenic viruses in clinical samples and provide real time data for a pathogens presence in a population.[28]
See also
References
- 1 2 3 4 Sommers P, Chatterjee A, Varsani A, Trubl G (September 2021). "Integrating Viral Metagenomics into an Ecological Framework". Annual Review of Virology. 8 (1): 133–158. doi:10.1146/annurev-virology-010421-053015. PMID 34033501.
- ↑ Grasis JA (2018). "Host-Associated Bacteriophage Isolation and Preparation for Viral Metagenomics". Viral Metagenomics. Methods in Molecular Biology. Vol. 1746. New York, NY: Springer New York. pp. 1–25. doi:10.1007/978-1-4939-7683-6_1. ISBN 978-1-4939-7682-9. PMID 29492882. S2CID 3637163. Retrieved 2022-12-02.
- 1 2 3 4 5 6 7 Krishnamurthy SR, Wang D (July 2017). "Origins and challenges of viral dark matter". Virus Research. 239: 136–142. doi:10.1016/j.virusres.2017.02.002. PMID 28192164.
- 1 2 3 4 5 6 7 Pappas N, Roux S, Hölzer M, Lamkiewicz K, Mock F, Marz M, Dutilh BE (2021). "Virus Bioinformatics". In Bamford DH, Zuckerman M (eds.). Encyclopedia of Virology (4th ed.). Elsevier. pp. 124–132. doi:10.1016/b978-0-12-814515-9.00034-5. ISBN 978-0-12-814516-6. PMC 7567488.
- ↑ Kristensen DM, Mushegian AR, Dolja VV, Koonin EV (January 2010). "New dimensions of the virus world discovered through metagenomics". Trends in Microbiology. 18 (1): 11–19. doi:10.1016/j.tim.2009.11.003. PMC 3293453. PMID 19942437.
- ↑ Bernardo P, Albina E, Eloit M, Roumagnac P (May 2013). "[Pathology and viral metagenomics, a recent history]". Medecine Sciences (in French). 29 (5): 501–508. doi:10.1051/medsci/2013295013. PMID 23732099.
- 1 2 Alavandi SV, Poornima M (September 2012). "Viral metagenomics: a tool for virus discovery and diversity in aquaculture". Indian Journal of Virology. 23 (2): 88–98. doi:10.1007/s13337-012-0075-2. PMC 3550753. PMID 23997432.
- 1 2 3 4 5 Santiago-Rodriguez, Tasha M.; Hollister, Emily B. (2022-09-16). "Unraveling the viral dark matter through viral metagenomics". Frontiers in Immunology. 13. doi:10.3389/fimmu.2022.1005107. ISSN 1664-3224. PMC 9523745. PMID 36189246.
- 1 2 3 4 5 6 7 Houldcroft CJ, Beale MA, Breuer J (March 2017). "Clinical and biological insights from viral genome sequencing". Nature Reviews. Microbiology. 15 (3): 183–192. doi:10.1038/nrmicro.2016.182. PMC 7097211. PMID 28090077.
- 1 2 Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM, Mead D, et al. (October 2002). "Genomic analysis of uncultured marine viral communities". Proceedings of the National Academy of Sciences of the United States of America. 99 (22): 14250–14255. Bibcode:2002PNAS...9914250B. doi:10.1073/pnas.202488399. PMC 137870. PMID 12384570.
- ↑ Simmonds P, Adams MJ, Benkő M, Breitbart M, Brister JR, Carstens EB, et al. (March 2017). "Consensus statement: Virus taxonomy in the age of metagenomics". Nature Reviews. Microbiology. 15 (3): 161–168. doi:10.1038/nrmicro.2016.177. PMID 28134265. S2CID 1478314.
- ↑ Koonin, Eugene V.; Krupovic, Mart; Agol, Vadim I. (2021-08-18). "The Baltimore Classification of Viruses 50 Years Later: How Does It Stand in the Light of Virus Evolution?". Microbiology and Molecular Biology Reviews. 85 (3): e0005321. doi:10.1128/MMBR.00053-21. ISSN 1092-2172. PMC 8483701. PMID 34259570.
- ↑ Depledge DP, Palser AL, Watson SJ, Lai IY, Gray ER, Grant P, et al. (2011-11-18). Jhaveri R (ed.). "Specific capture and whole-genome sequencing of viruses from clinical samples". PLOS ONE. 6 (11): e27805. Bibcode:2011PLoSO...627805D. doi:10.1371/journal.pone.0027805. PMC 3220689. PMID 22125625.
- 1 2 Thomson E, Ip CL, Badhan A, Christiansen MT, Adamson W, Ansari MA, et al. (October 2016). "Comparison of Next-Generation Sequencing Technologies for Comprehensive Assessment of Full-Length Hepatitis C Viral Genomes". Journal of Clinical Microbiology. 54 (10): 2470–2484. doi:10.1128/jcm.00330-16. PMC 5035407. PMID 27385709.
- ↑ Paez-Espino D, Chen IA, Palaniappan K, Ratner A, Chu K, Szeto E, et al. (January 2017). "IMG/VR: a database of cultured and uncultured DNA Viruses and retroviruses". Nucleic Acids Research. 45 (D1): D457–D465. doi:10.1093/nar/gkw1030. PMC 5210529. PMID 27799466.
- ↑ Paez-Espino D, Roux S, Chen IA, Palaniappan K, Ratner A, Chu K, et al. (January 2019). "IMG/VR v.2.0: an integrated data management and analysis system for cultivated and environmental viral genomes". Nucleic Acids Research. 47 (D1): D678–D686. doi:10.1093/nar/gky1127. PMC 6323928. PMID 30407573.
- ↑ Quick J (2019-09-25). "Ebola virus sequencing protocol v1". Protocols.io. doi:10.17504/protocols.io.7nwhmfe. S2CID 216572035. Retrieved 2022-11-30.
- ↑ Vlachakis D, Papageorgiou L, Megalooikonomou V (2018-06-13), "Genetic and Geo-Epidemiological Analysis of the Zika Virus Pandemic; Learning Lessons from the Recent Ebola Outbreak", Current Topics in Zika, InTech, doi:10.5772/intechopen.71505, ISBN 978-1-78923-270-7, S2CID 90434818
- ↑ Charre C, Ginevra C, Sabatier M, Regue H, Destras G, Brun S, et al. (July 2020). "Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation". Virus Evolution. 6 (2): veaa075. bioRxiv 10.1101/2020.07.14.201947. doi:10.1093/ve/veaa075. PMC 7665770. PMID 33318859.
- ↑ Ebert K, Depledge DP, Breuer J, Harman L, Elliott G (September 2013). "Mode of virus rescue determines the acquisition of VHS mutations in VP22-negative herpes simplex virus 1". Journal of Virology. 87 (18): 10389–10393. doi:10.1128/jvi.01654-13. PMC 3753997. PMID 23864617.
- ↑ Depledge DP, Palser AL, Watson SJ, Lai IY, Gray ER, Grant P, et al. (2011-11-18). "Specific capture and whole-genome sequencing of viruses from clinical samples". PLOS ONE. 6 (11): e27805. Bibcode:2011PLoSO...627805D. doi:10.1371/journal.pone.0027805. PMC 3220689. PMID 22125625.
- 1 2 García-López R, Vázquez-Castellanos JF, Moya A (September 2015). "Fragmentation and Coverage Variation in Viral Metagenome Assemblies, and Their Effect in Diversity Calculations". Frontiers in Bioengineering and Biotechnology. 3: 141. doi:10.3389/fbioe.2015.00141. PMC 4585024. PMID 26442255.
- 1 2 3 Arroyo Mühr LS, Lagheden C, Hassan SS, Kleppe SN, Hultin E, Dillner J (August 2020). "De novo sequence assembly requires bioinformatic checking of chimeric sequences". PLOS ONE. 15 (8): e0237455. Bibcode:2020PLoSO..1537455A. doi:10.1371/journal.pone.0237455. PMC 7417191. PMID 32777809.
- ↑ Brewer HC, Hird DL, Bailey AM, Seal SE, Foster GD (April 2018). "A guide to the contained use of plant virus infectious clones". Plant Biotechnology Journal. 16 (4): 832–843. doi:10.1111/pbi.12876. PMC 5867029. PMID 29271098.
- ↑ Pasin F, Menzel W, Daròs JA (June 2019). "Harnessed viruses in the age of metagenomics and synthetic biology: an update on infectious clone assembly and biotechnologies of plant viruses". Plant Biotechnology Journal. 17 (6): 1010–1026. doi:10.1111/pbi.13084. PMC 6523588. PMID 30677208.
- ↑ Pratama AA, van Elsas JD (August 2018). "The 'Neglected' Soil Virome - Potential Role and Impact". Trends in Microbiology. 26 (8): 649–662. doi:10.1016/j.tim.2017.12.004. PMID 29306554. S2CID 25057850.
- 1 2 Schmidt C (October 2018). "The virome hunters". Nature Biotechnology. 36 (10): 916–919. doi:10.1038/nbt.4268. PMC 7097093. PMID 30307913.
- 1 2 Roux S, Matthijnssens J, Dutilh BE (2021), "Metagenomics in Virology", Encyclopedia of Virology, Elsevier, pp. 133–140, doi:10.1016/b978-0-12-809633-8.20957-6, ISBN 978-0-12-814516-6, PMC 7157462
- ↑ Vernimmen T (2020-04-16). "Infectious disease: Making — and breaking — the animal connection". Knowable Magazine | Annual Reviews. doi:10.1146/knowable-041620-1. S2CID 218810265.
- ↑ "Contact". Global Virome Project. Archived from the original on 2022-08-22. Retrieved 2022-08-22.
- 1 2 Dutilh BE, Reyes A, Hall RJ, Whiteson KL (September 2017). "Editorial: Virus Discovery by Metagenomics: The (Im)possibilities". Frontiers in Microbiology. 8 (1710): 1710. doi:10.3389/fmicb.2017.01710. PMC 5596103. PMID 28943867.
External links
- IMG/VR The IMG Viral Database (IMG/VR).
- CAMERA Archived 2012-05-14 at the Wayback Machine Cyberinfrastructure for Metagenomics, data repository and tools for metagenomics research.
- GOLD Genomes OnLine Database (GOLD).
- IMG/M The Integrated Microbial Genomes system, for metagenome analysis by the DOE-JGI.
- MEGAN Archived 2010-05-07 at the Wayback Machine MEtaGenome ANalyzer. A stand-alone metagenome analysis tool.
- MetaGeneMark MetaGeneMark for MetaGenome Gene Finding
- Metagenomics and Our Microbial Planet Archived 2014-03-26 at the Wayback Machine A website on metagenomics and the vital role of microbes on Earth from the National Academies.
- Metagenomics at the European Bioinformatics Institute Analysis and archiving of metagenomic data.
- Metagenomics: Sequences from the Environment free ebook from NCBI Bookshelf.
- MG-RAST Metagenomics Rapid Annotation using Subsystem Technology server
- The New Science of Metagenomics: Revealing the Secrets of Our Microbial Planet A report released by the National Research Council in March 2007. Also, see the Report In Brief. Archived 2010-04-15 at the Wayback Machine
- http://www.globalviromeproject.org/ Official site of the Global Virome Project
- https://www.usaid.gov/ept2 Emerging pandemic threats program EPT-2
- https://www.ecohealthalliance.org/program/predict
| Genomics | |
|---|---|
| Bioinformatics | |
| Structural biology | |
| Research tools | |
| Organizations | |
| |
| Components |  | |
|---|---|---|
| Viral life cycle | ||
| Genetics | ||
| By host | ||
| Other | ||