Dynamic kinetic resolution in chemistry is a type of kinetic resolution where 100% of a racemic compound can be converted into an enantiopure compound. It is applied in asymmetric synthesis. Asymmetric synthesis has become a much explored field due to the challenge of creating a compound with a single 3D structure.[1] Even more challenging is the ability to take a racemic mixture and have only one chiral product left after a reaction. One method that has become an exceedingly useful tool is dynamic kinetic resolution (DKR).[2][3] DKR utilizes a center of a particular molecule that can be easily epimerized so that the (R) and (S) enantiomers can interconvert throughout the reaction process. At this point the catalyst can selectively lower the transition state energy of a single enantiomer, leading to almost 100% yield of one reaction pathway over the other. The figure below is an example of an energy diagram for a compound with an (R) and (S) isomer.[4]

If a catalyst is able to increase ΔΔG‡ to a sufficient degree, then one pathway will dominate over the other, leading to a single chiral product. Manipulating kinetics therefore becomes a powerful way to achieve asymmetric products from racemic starting materials. There have been numerous uses of DKR in the literature that have provided new methods in pharmaceuticals[5] as well as routes to natural products.[6]
Applications
Noyori's asymmetric hydrogenation
One of the more classic applications of DKR is Noyori's asymmetric hydrogenation.[7] The presence of an acidic center between two carbonyl groups allows for easy epimerization at the chiral center under basic conditions. To select for one of the four possible stereoisomers, a BINAP-Ru catalyst is used to control the outcome of the reaction through the steric bulk of the phosphorus ligand. Some of the early transformations are shown below.

To further understand the stereochemical outcome, one must look at the transition state geometry.

The steric bulk of the BINAP ligand coupled with the coordination of ruthenium to the carbonyl oxygen atoms results in high selectivity for hydrogen insertion on one face. This resulting stereochemistry of (R,S) and (R,R) is obtained in 94.5% yield while the other three stereoisomers range from 0.5-3% yield. Noyori's accomplishments of 1990 paved the way for even more useful applications of DKR.
Asymmetric conjugate reduction
About a decade later, Jurkauskas and Buchwald also utilized dynamic kinetic resolution towards the hydrogenation of conjugated systems.[8] 1,4 addition to cyclic enones is quite common in many reaction schemes, however asymmetric reductions in the presence of an easily epimerizable center adds to the complexity when trying to modify only one center. Through the use of a copper catalyzed reaction however, Buchwald was able to obtain 1,4 reduction in great enantiomeric excess (ee). In order to achieve a high rate of epimerization, a strong bulky base like sodium t-butoxide was used to ensure rapid equilibrium.

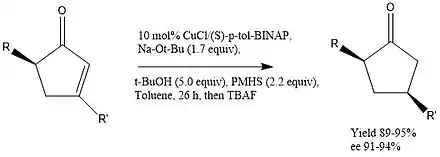
Copper proved to be an excellent metal in this reaction due to its ability to complex with the oxygen when the hydrogen was added. Being a soft metal, copper greatly prefers 1,4 addition over 1,2 addition, with the alkene being a softer more polarizable electrophile. Again, BINAP became the ligand of choice due to its steric selectivity, lowering the transition state energy of starting material in the left column. In addition, PMHS was used as a relatively less reactive silane. This prevented loss of ee before deprotection with tetra-n-butylammonium fluoride (TBAF).
Asymmetric aldol reaction
In addition to hydrogenation reactions, other bonds have been formed using DKR and are highly successful.[9][10][11] The aldol reaction has been extensively researched primarily because of the inherent challenge of forming a carbon-carbon bond.[12][13] Ward and colleagues have been able to use the proline-catalyzed aldol reaction in tandem with dynamic kinetic resolution to obtain a high enantioselective reaction.[14]
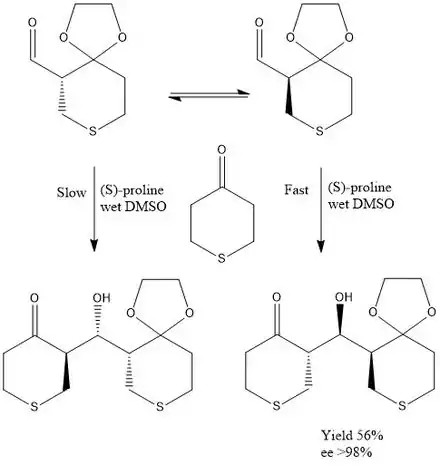
In this reaction proline catalyzes the reaction through creation of an enamine intermediate that is highly nucleophilic. The acid group on the catalyst helps facilitate the carbon-carbon bond formation by coordinating with the aldehyde oxygen. This greatly improves stereoselectivity and yield. Ward and his associates also found that by adding trace amounts of water to the DMSO solvent, it greatly increase the yield of the reaction, most likely by aiding proton transfer from proline to the newly forming alcohol.

The selectivity for this product can best be explained by the Felkin model. The cyclic (E)-enamine is able to undergo a favorable transition state where the aldehyde adopts an anti relationship relative to the incoming nucleophile, as well as a 1,2 syn relationship between the aldehyde and its adjacent ring system. The transition state is shown above.
Enzyme-metal reactions
More recently many research groups have tried to employ enzymes into DKR synthetic routes.[15][16] Due to the generally high specificity for substrates, enzymes prove to be vital catalysts for binding to only one stereoisomer in the racemic mixture. In 2007 Bäckvall discovered an enzyme-metal coupled reaction that converts allylic acetates to allylic alcohols with excellent stereospecificity.[17]
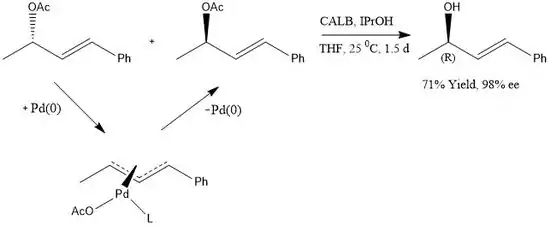
In this reaction, a Pd(0) complex is used to interconvert the chirality of the acetate center at a rate fast enough to ensure complete racemization. When this is achieved the CALB enzyme selectively hydrolyzes the (R) substrate because of the low binding affinity for the (S) substrate. This gives almost exclusively the (R) allylic alcohol in 98% ee.
To expand on this chemistry, Bäckvall designed a one-pot, two-reaction system that utilizes the stereochemical outcome of a DKR reaction to undergo a second energetically favorable reaction with high enantioselectivity.

This time a ruthenium complex is used to racemize the allylic alcohol in much the same way as the previous example. The addition of CALB catalyzes the reaction between the (R) isomer and the ester reagent to form a product with a diene and a dienophile. This intermediate can then undergo a tandem Diels-Alder reaction to achieve a decent yield with 97% ee.
Natural product synthesis
Dynamic kinetic resolution has also been applied to the total synthesis of a variety of natural products. After Bäckvall's discoveries in 2007, he employed another enzyme-metal coupled reaction to synthesize the natural product (R)-Bufuralol.[18]

The key step that the literature points out utilizes DKR to convert the chlorohydrin into the (S)-acetate by means of a lipase and a ruthenium catalyst.

The lipase PS-C “Amano” II has been reported in the literature to be particularly enantioselective for the 1-phenyl-2-chloroethanol motif. The enzyme, along with the ruthenium catalyst, allows for rapid racemization of the chlorohydrin with a selective binding to the (S) isomer for the acetylation reaction. Here isopropenyl acetate is used as the acyl donor. The product is achieved in excellent yield (96%) and near-perfect enantiomeric excess (>99%).
Conclusion
With the number of asymmetric synthetic challenges increasing as new targets for pharmaceuticals and materials grow, methods development becomes critical.[19] Dynamic kinetic resolution is one solution to this ever growing demand, as one can take inexpensive racemic starting materials and come out with products in high yield and stereoselectivity.[20] As the scope and application of this powerful concept increases, its utilization in the industrial and academic settings is likely to expand in the years to come.
References
- ↑ El, G. M. T.; Williams, J. M. J. Curr. Opin. Chem. Biol. 1999, 3, 11–15.
- ↑ Pellissier, H. Tetrahedron 2008, 64, 1563–1601.
- ↑ Coldham, I.; Dufour, S.; Haxell, T. F. N.; Patel, J. J.; Sanchez-Jimenez, G. J. Am. Chem. Soc. 2006, 128, 10943–10951.
- ↑ Hanefeld, U.; Veum, L. Tetrahedron: Asymmetry, 2004, 15, 3707–3709.
- ↑ Blacker, J.; Headley, C. E. In Green Chemistry in the Pharmaceutical Industry; 2010; pp. 269–288.
- ↑ Goodyear, M. D.; Hill, M. L.; West, J. P.; Whitehead, A. J. Tetrahedron Lett. 2005, 46, 8535–8538.
- ↑ Noyori, R.; Tokunaga, M.; Kitamura, M.; Ohkuma, T. Tetrahedron: Asymmetry, 1990, 1, 1–4.
- ↑ Jurkauskas, V.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 2892–2893.
- ↑ Hayakawa, Y.; Hyodo, M.; Kimura, K.; Kataoka, M. Chem. Commun. 2003, 1704–1705.
- ↑ Hoffmann, S.; Nicoletti, M.; List, B. J. Am. Chem. Soc. 2006, 128,13074–13075.
- ↑ Makino, K.; Iwasaki, M.; Hamada, Y. Org. Lett. 2006, 8, 4573–4576.
- ↑ Pàmies, O.; Bäckvall, J.-E. J. Org. Chem. 2002, 67, 9006–9010.
- ↑ Päiviö, M.; Mavrynsky, D.; Leino, R.; Kanerva, L. T. European J. Org. Chem. 2011, 2011, 1452–1457.
- ↑ Ward, D. E.; Jheengut, V.; Akinnusi, O. T. Org. Lett. 2005, 7, 1181–1184.
- ↑ Kim, M. J.; Ahn, Y.; Park, J. Curr. Opin. Biotechnol. 2002, 13, 578–587.
- ↑ Huerta, F.; Minidis, A. Chem. Soc. Rev. 2001, 30, 321–331.
- ↑ Martín-Matute, B.; Bäckvall, J.-E. Curr. Opin. Chem. Biol. 2007, 11, 226–232.
- ↑ Johnston, E. V; Bogár, K.; Bäckvall, J.-E. J. Org. Chem. 2010, 75, 4596–4599.
- ↑ Han, J.; Kang, S.; Lee, H. K. Chem. Commun. 2011, 47, 4004–4006.
- ↑ Ito, T.; Overman, L. E.; Wang, J. J. Am. Chem. Soc. 2010, 132, 3272–3273.