| Galactosemia | |
|---|---|
 | |
| Galactose | |
| Specialty | Endocrinology |
Galactosemia (British galactosaemia, from Greek γαλακτόζη + αίμα, meaning galactose + blood, accumulation of galactose in blood) is a rare genetic metabolic disorder that affects an individual's ability to metabolize the sugar galactose properly. Galactosemia follows an autosomal recessive mode of inheritance that confers a deficiency in an enzyme responsible for adequate galactose degradation.
Friedrich Goppert (1870–1927), a German physician, first described the disease in 1917,[1] with its cause as a defect in galactose metabolism being identified by a group led by Herman Kalckar in 1956.[2] Galactosemia was the second disorder found to be detectable through newborn screening methods by Robert Guthrie.[3]
Its incidence is about 1 per 60,000 births for people of European ancestry. In other populations the incidence rate differs. Galactosaemia is about one hundred times more common (1:480 births)[4] in the Irish Traveller population.[5]
Symptoms
Adults
Infants
Infants may appear asymptomatic at birth, however, upon ingestion of galactose a few days later (via breast and/or formula feeding), children start to experience life-threatening symptoms, which include:[6]
- poor feeding, and weight gain
- vomiting and diarrhea
- hepatocellular damage
- lethargy, and hypotonia
Progression of this acute neonatal toxicity syndrome may include the development of sepsis, cataracts, and even pseudotumor cerebri (which may cause a bulging of fontanelle).[6]
Children
In children, untreated galactosemia can lead to cataracts, developmental delays, intellectual disabilities, speech difficulties, fine and gross motor difficulties, kidney disease, liver failure, sepsis, and premature ovarian insufficiency. While a child is being treated for galactosemia, they may continue to experience speech delays, learning disabilities, behavioral issues, ataxia, tremors, and hormone deficiencies.[7]
Cause

Lactose in food (such as dairy products) is broken down by the enzyme lactase into glucose and galactose. In individuals with galactosemia, the enzymes needed for further metabolism of galactose (Galactokinase and galactose-1-phosphate uridyltransferase) are severely diminished or missing entirely, leading to toxic levels of galactose or galactose 1-phosphate (depending on which enzyme is missing) in various tissues as in the case of classic galactosemia, resulting in hepatomegaly (an enlarged liver), cirrhosis, kidney failure, cataracts, vomiting, seizure, low blood sugar (hypoglycemia), lethargy, brain damage, and ovarian failure. Without treatment, mortality in infants with galactosemia is about 75%.[8]
Galactosemia is inherited in an autosomal recessive manner, meaning a child must inherit one defective gene from each parent to show the disease. Heterozygotes are carriers, because they inherit one normal gene and one defective gene.[9] Carriers show no symptoms of galactosemia.[10]
Accumulation of galactose
Reduction to galactitol
In galactosemic patients, the accumulation of galactose becomes the substrate for enzymes that catalyze the polyol pathway of carbohydrate metabolism. The first reaction of this pathway is the reduction of aldoses, types of sugars including galactose, to sugar alcohols.[11] Recent data suggests that aldose reductase is the enzyme responsible for the primary stage of this pathway. Therefore, aldose reductase reduces galactose to its sugar alcohol form, galactitol. Galactitol, however, is not a suitable substrate for the next enzyme in the polyol pathway, polyol dehydrogenase. Thus, galactitol accumulates in body tissues and is excreted in the urine of galactosemic patients. Accumulation of galactitol has been attributed to many of the negative effects of galactosemia, and high concentrations of galactitol have been found in people with classic galactosemia (GALT deficiency or Galactose-1-phosphate uridylyltransferase deficiency), galactokinase deficiency, and epimerase deficiency with glucose.
Oxidation to galactonate
Accumulated galactose can also undergo an alternative reaction: Oxidation to galactonate. The mechanism of galactonate formation is still unclear. However, recent studies suggest that galactose dehydrogenase is responsible for converting galactose to galactonolactone, which then spontaneously or enzymatically converts to galactonate. Once formed, galactonate may enter the pentose phosphate pathway. Thus, oxidation to galactonate serves as an alternate pathway for metabolizing galactose. This oxidative pathway renders accumulated galactonate less harmful than accumulated galactitol.
Diagnosis
In many states throughout the world, infants routinely undergo newborn screening (NBS) for galactosemia.[6] This allows a diagnosis to be made while the person is still an infant. Infants affected by galactosemia typically present with symptoms of lethargy, vomiting, diarrhea, failure to thrive, and jaundice. None of these symptoms are specific to galactosemia, often leading to diagnostic delays. If the family of the baby has a history of galactosemia, doctors can test prior to birth by taking a sample of fluid from around the fetus (amniocentesis) or from the placenta (chorionic villus sampling or CVS).[12] Galactosemia is normally first detected through newborn screening which if available, is able to diagnose the majority of affected infants.
A galactosemia test is a blood test (from the heel of the infant) or urine test that checks for three enzymes that are needed to change galactose sugar that is found in milk and milk products into glucose, a sugar that the human body uses for energy. A person with galactosemia doesn't have one of these enzymes. This causes high levels of galactose in the blood or urine.[13][14]
Affected children can have serious, irreversible effects or even die within days from birth. It is important that newborns be screened for metabolic disorders without delay. Galactosemia can even be detected through NBS before any ingestion of galactose-containing formula or breast milk.
Detection of the disorder through NBS does not depend on protein or lactose ingestion, and, therefore, it should be identified on the first specimen unless the infant has been transfused. A specimen should be taken prior to transfusion. The enzyme is prone to damage if analysis of the sample is delayed or exposed to high temperatures. The routine NBS is accurate for detection of galactosemia. Two screening tests are used to screen infants affected with galactosemia—the Beutler's test and the Hill test.[15] The Beutler's test screens for galactosemia by detecting the level of enzyme of the infant. Therefore, the ingestion of formula or breast milk does not affect the outcome of this part of the NBS, and the NBS is accurate for detecting galactosemia prior to any ingestion of galactose.
Duarte galactosemia is a milder form of classical galactosemia and usually has no long term side effects.[16]
Types
Galactose is converted into glucose by the action of three enzymes, known as the Leloir pathway. There are diseases associated with deficiencies of each of these three enzymes:
| Type | Diseases Database | OMIM | Gene | Locus | Enzyme | Name |
| Type 1 | 5056 | 230400 | GALT | 9p13 | galactose-1-phosphate uridyl transferase | classic galactosemia |
| Type 2 | 29829 | 230200 | GALK1 | 17q24 | galactokinase | galactokinase deficiency |
| Type 3 | 29842 | 230350 | GALE | 1p36-p35 | UDP galactose epimerase | galactose epimerase deficiency, UDP-Galactose-4-epimerase deficiency |
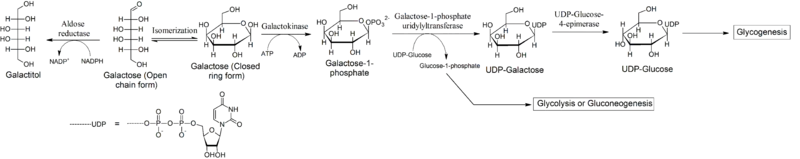

Treatment
The only treatment for classic galactosemia is eliminating lactose and galactose from the diet (e.g. exclusion of dairy products containing lactose).[6][17][18]
Lactose restricted diet is efficient in resolving acute complications, however, it is not sufficient to prevent long-term complications affecting the brain and female gonads.[17] Some individuals may experience long-term complications such as speech difficulties, learning disabilities, neurological impairment (e.g. tremors, etc.), and ovarian failure.[19]
Symptoms that have not been associated with Duarte galactosemia,[20] and many individuals with Duarte galactosemia, do not need to restrict their diet at all. However, research corroborates a previously overlooked theory that Duarte galactosemia may lead to language developmental issues in children with no clinical symptoms. Infants with classic galactosemia cannot be breast-fed due to lactose in human breast milk which consists of both galactose and glucose and are usually fed a soy-based formula.[21]
Galactosemia is sometimes confused with lactose intolerance, but galactosemia is a more serious condition. Lactose intolerant individuals have an acquired or inherited shortage of the enzyme lactase, and experience abdominal pains after ingesting dairy products, but no long-term effects. In contrast, a galactosemic individual who consumes galactose can have serious complications including:
- Speech deficits[22]
- Ataxia
- Dysmetria
- Diminished bone density
- Premature ovarian failure[23]
- Cataract
See also
References
- ↑ Goppert F. (1917). "Galaktosurie nach Milchzuckergabe bei angeborenem, familiaerem chronischem Leberleiden". Klinische Wochenschrift. 54: 473–7.
- ↑ Isselbacher KJ, Anderson EP, Kurahashi K, Kalckar HM (1956). "Congenital Galactosemia, a single enzymatic block in galactose metabolism". Science. 123 (3198): 635–6. Bibcode:1956Sci...123..635I. doi:10.1126/science.123.3198.635. PMID 13311516.
- ↑ "History of Galactosemia". Galactosemia Foundation. Archived from the original on 2021-04-25. Retrieved 2020-11-05.
- ↑ "Classical Galactosaemia - Ireland's Health Service". www.hse.ie.
- ↑ Murphy M, McHugh B, Tighe O, et al. (July 1999). "Genetic basis of transferase-deficient Galactosaemia in Ireland and the population history of the Irish Travellers". Eur. J. Hum. Genet. 7 (5): 549–54. doi:10.1038/sj.ejhg.5200327. PMID 10439960.
- 1 2 3 4 Coelho AI, Rubio-Gozalbo ME, Vicente JB, Rivera I (May 2017). "Sweet and sour: an update on classic galactosemia". Journal of Inherited Metabolic Disease. 40 (3): 325–342. doi:10.1007/s10545-017-0029-3. PMC 5391384. PMID 28281081.
- ↑ "Galactosemia". Cleveland Clinic. Retrieved 17 February 2023.
- ↑ "Genetics Laboratory - The University of Oklahoma Health Sciences Center". genetics.ouhsc.edu. Retrieved 2023-07-26.
- ↑ Galactosemia Archived April 19, 2016, at the Wayback Machine The University of Utah, Genetics Science Learning Center. 2008.
- ↑ "Galactosemia - Symptoms, Causes, Treatment | NORD". rarediseases.org. Retrieved 2023-07-26.
- ↑ Kolatkar, Nikheel Dr. "Aldose Rudctase Inhibitors." Your Total Health. "IVillage Total Health > Health Guide Detail". Archived from the original on 2009-07-12. Retrieved 2011-01-19.
- ↑ Fensom AH, Benson PF, Blunt S (November 1974). "Prenatal diagnosis of galactosaemia". Br Med J. 4 (5941): 386–7. doi:10.1136/bmj.4.5941.386. PMC 1612460. PMID 4154122.
- ↑ Berry, Gerard T. (1993), Adam, Margaret P.; Mirzaa, Ghayda M.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Classic Galactosemia and Clinical Variant Galactosemia", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 20301691, retrieved 2023-07-26
- ↑ Palmieri, M.; Mazur, A.; Berry, G. T.; Ning, C.; Wehrli, S.; Yager, C.; Reynolds, R.; Singh, R.; Muralidharan, K.; Langley, S.; Elsas, L.; Segal, S. (October 1999). "Urine and plasma galactitol in patients with galactose-1-phosphate uridyltransferase deficiency galactosemia". Metabolism: Clinical and Experimental. 48 (10): 1294–1302. doi:10.1016/s0026-0495(99)90271-8. ISSN 0026-0495. PMID 10535394.
- ↑ Parris CR (August 2006). "An Overview of Expanded Newborn Screening for Inborn Errors of Metabolism" (PDF). Nutrition Issues in Gastroenterology. Archived from the original (PDF) on 2015-09-20.
- ↑ Duarte galactosemia (DG or D/G galactosemia), Minnesota Department of Health
- 1 2 Demirbas, Didem; Coelho, Ana I.; Rubio-Gozalbo, M. Estela; Berry, Gerard T. (June 2018). "Hereditary galactosemia". Metabolism: Clinical and Experimental. 83: 188–196. doi:10.1016/j.metabol.2018.01.025. ISSN 1532-8600. PMID 29409891. S2CID 29149432.
- ↑ Kiss, Erika; Balogh, Lídia; Reismann, Péter (November 2017). "[Diet treatment of classical galactosemia]". Orvosi Hetilap. 158 (47): 1864–1867. doi:10.1556/650.2017.30900. ISSN 0030-6002. PMID 29153024.
- ↑ Fridovich-Keil, Judith L. (April 2011). "Ovarian function in girls and women with GALT-deficiency galactosemia". J Inherit Metab Dis. 34 (2): 357–66. doi:10.1007/s10545-010-9221-4. PMC 3063539. PMID 20978943.
- ↑ "GARD Rare Disease Information - Duarte Galactosemia - National Organization for Rare Disorders". rarediseases.org. 2022-06-16. Retrieved 2023-07-26.
- ↑ "Breastfeeding: Diseases and Conditions: Contraindicators | DNPA". www.cdc.gov.
- ↑ Powell KK, Van Naarden Braun K, Singh RH, Shapira SK, Olney RS, Yeargin-Allsopp M (2009), "Long-term speech and language developmental issues among children with Duarte galactosemia.", Genetics in Medicine, 11 (12): 874–9, doi:10.1097/GIM.0b013e3181c0c38d, PMID 19904210
- ↑ Waggoner DD, Buist NR, Donnell GN (1990), "Long-term prognosis in galactosaemia: results of a survey of 350 cases..", J Inherit Metab Dis, 13 (6): 802–18, doi:10.1007/BF01800204, PMID 1706789, S2CID 19083275