ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
Uses
ChIP-seq is primarily used to determine how transcription factors and other chromatin-associated proteins influence phenotype-affecting mechanisms. Determining how proteins interact with DNA to regulate gene expression is essential for fully understanding many biological processes and disease states. This epigenetic information is complementary to genotype and expression analysis. ChIP-seq technology is currently seen primarily as an alternative to ChIP-chip which requires a hybridization array. This introduces some bias, as an array is restricted to a fixed number of probes. Sequencing, by contrast, is thought to have less bias, although the sequencing bias of different sequencing technologies is not yet fully understood.[1]
Specific DNA sites in direct physical interaction with transcription factors and other proteins can be isolated by chromatin immunoprecipitation. ChIP produces a library of target DNA sites bound to a protein of interest. Massively parallel sequence analyses are used in conjunction with whole-genome sequence databases to analyze the interaction pattern of any protein with DNA,[2] or the pattern of any epigenetic chromatin modifications. This can be applied to the set of ChIP-able proteins and modifications, such as transcription factors, polymerases and transcriptional machinery, structural proteins, protein modifications, and DNA modifications.[3] As an alternative to the dependence on specific antibodies, different methods have been developed to find the superset of all nucleosome-depleted or nucleosome-disrupted active regulatory regions in the genome, like DNase-Seq[4] and FAIRE-Seq.[5][6]
Workflow of ChIP-sequencing
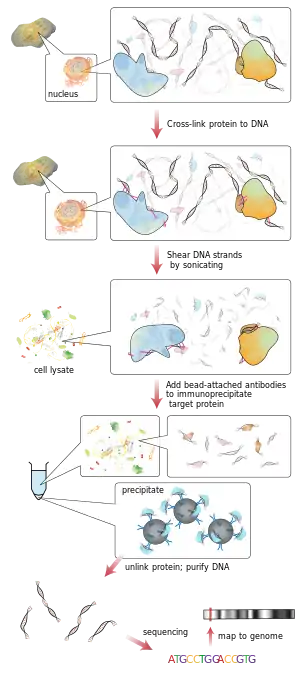
ChIP
ChIP is a powerful method to selectively enrich for DNA sequences bound by a particular protein in living cells. However, the widespread use of this method has been limited by the lack of a sufficiently robust method to identify all of the enriched DNA sequences. The ChIP wet lab protocol contains ChIP and hybridization. There are essentially five parts to the ChIP protocol[7] that aid in better understanding the overall process of ChIP. In order to carry out the ChIP, the first step is cross-linking[8] using formaldehyde and large batches of the DNA in order to obtain a useful amount. The cross-links are made between the protein and DNA, but also between RNA and other proteins. The second step is the process of chromatin fragmentation which breaks up the chromatin in order to get high quality DNA pieces for ChIP analysis in the end. These fragments should be cut to become under 500 base pairs[9] each to have the best outcome for genome mapping. The third step is called chromatin immunoprecipitation,[7] which is what ChIP is short for. The ChIP process enhances specific crosslinked DNA-protein complexes using an antibody against the protein of interest followed by incubation and centrifugation to obtain the immunoprecipitation. The immunoprecipitation step also allows for the removal of non-specific binding sites. The fourth step is DNA recovery and purification,[7] taking place by the reversed effect on the cross-link between DNA and protein to separate them and cleaning DNA with an extraction. The fifth and final step is the analyzation step of the ChIP protocol by the process of qPCR, ChIP-on-chip (hybrid array) or ChIP sequencing. Oligonucleotide adaptors are then added to the small stretches of DNA that were bound to the protein of interest to enable massively parallel sequencing. Through the analysis, the sequences can then be identified and interpreted by the gene or region to where the protein was bound.[7]
Sequencing
After size selection, all the resulting ChIP-DNA fragments are sequenced simultaneously using a genome sequencer. A single sequencing run can scan for genome-wide associations with high resolution, meaning that features can be located precisely on the chromosomes. ChIP-chip, by contrast, requires large sets of tiling arrays for lower resolution.[10]
There are many new sequencing methods used in this sequencing step. Some technologies that analyze the sequences can use cluster amplification of adapter-ligated ChIP DNA fragments on a solid flow cell substrate to create clusters of approximately 1000 clonal copies each. The resulting high density array of template clusters on the flow cell surface is sequenced by a genome analyzing program. Each template cluster undergoes sequencing-by-synthesis in parallel using novel fluorescently labelled reversible terminator nucleotides. Templates are sequenced base-by-base during each read. Then, the data collection and analysis software aligns sample sequences to a known genomic sequence to identify the ChIP-DNA fragments.
Quality control
ChIP-seq offers us a fast analysis, however, a quality control must be performed to make sure that the results obtained are reliable:
Sensitivity
Sensitivity of this technology depends on the depth of the sequencing run (i.e. the number of mapped sequence tags), the size of the genome and the distribution of the target factor. The sequencing depth is directly correlated with cost. If abundant binders in large genomes have to be mapped with high sensitivity, costs are high as an enormously high number of sequence tags will be required. This is in contrast to ChIP-chip in which the costs are not correlated with sensitivity.[12][13]
Unlike microarray-based ChIP methods, the precision of the ChIP-seq assay is not limited by the spacing of predetermined probes. By integrating a large number of short reads, highly precise binding site localization is obtained. Compared to ChIP-chip, ChIP-seq data can be used to locate the binding site within few tens of base pairs of the actual protein binding site. Tag densities at the binding sites are a good indicator of protein–DNA binding affinity,[14] which makes it easier to quantify and compare binding affinities of a protein to different DNA sites.[15]
Current research
STAT1 DNA association: ChIP-seq was used to study STAT1 targets in HeLa S3 cells which are clones of the HeLa line that are used for analysis of cell populations.[16] The performance of ChIP-seq was then compared to the alternative protein–DNA interaction methods of ChIP-PCR and ChIP-chip.[17]
Nucleosome Architecture of Promoters: Using ChIP-seq, it was determined that Yeast genes seem to have a minimal nucleosome-free promoter region of 150bp in which RNA polymerase can initiate transcription.[18]
Transcription factor conservation: ChIP-seq was used to compare conservation of TFs in the forebrain and heart tissue in embryonic mice. The authors identified and validated the heart functionality of transcription enhancers, and determined that transcription enhancers for the heart are less conserved than those for the forebrain during the same developmental stage.[19]
Genome-wide ChIP-seq: ChIP-sequencing was completed on the worm C. elegans to explore genome-wide binding sites of 22 transcription factors. Up to 20% of the annotated candidate genes were assigned to transcription factors. Several transcription factors were assigned to non-coding RNA regions and may be subject to developmental or environmental variables. The functions of some of the transcription factors were also identified. Some of the transcription factors regulate genes that control other transcription factors. These genes are not regulated by other factors. Most transcription factors serve as both targets and regulators of other factors, demonstrating a network of regulation.[20]
Inferring regulatory network: ChIP-seq signal of Histone modification were shown to be more correlated with transcription factor motifs at promoters in comparison to RNA level.[21] Hence author proposed that using histone modification ChIP-seq would provide more reliable inference of gene-regulatory networks in comparison to other methods based on expression.
ChIP-seq offers an alternative to ChIP-chip. STAT1 experimental ChIP-seq data have a high degree of similarity to results obtained by ChIP-chip for the same type of experiment, with greater than 64% of peaks in shared genomic regions. Because the data are sequence reads, ChIP-seq offers a rapid analysis pipeline as long as a high-quality genome sequence is available for read mapping and the genome doesn't have repetitive content that confuses the mapping process. ChIP-seq also has the potential to detect mutations in binding-site sequences, which may directly support any observed changes in protein binding and gene regulation.
Computational analysis
As with many high-throughput sequencing approaches, ChIP-seq generates extremely large data sets, for which appropriate computational analysis methods are required. To predict DNA-binding sites from ChIP-seq read count data, peak calling methods have been developed. One of the most popular methods is MACS which empirically models the shift size of ChIP-Seq tags, and uses it to improve the spatial resolution of predicted binding sites.[22] MACS is optimized for higher resolution peaks, while another popular algorithm, SICER is programmed to call for broader peaks, spanning over kilobases to megabases in order to search for broader chromatin domains. SICER is more useful for histone marks spanning gene bodies. A mathematical more rigorous method BCP (Bayesian Change Point) can be used for both sharp and broad peaks with faster computational speed,[23] see benchmark comparison of ChIP-seq peak-calling tools by Thomas et al. (2017).[24]
Another relevant computational problem is differential peak calling, which identifies significant differences in two ChIP-seq signals from distinct biological conditions. Differential peak callers segment two ChIP-seq signals and identify differential peaks using Hidden Markov Models. Examples for two-stage differential peak callers are ChIPDiff[25] and ODIN.[26]
To reduce spurious sites from ChIP-seq, multiple experimental controls can be used to detect binding sites from an IP experiment. Bay2Ctrls adopts a Bayesian model to integrate the DNA input control for the IP, the mock IP and its corresponding DNA input control to predict binding sites from the IP.[27] This approach is particularly effective for complex samples such as whole model organisms. In addition, the analysis indicates that for complex samples mock IP controls substantially outperform DNA input controls probably due to the active genomes of the samples.[27]
See also
Similar methods
- CUT&RUN sequencing, antibody-targeted controlled cleavage by micrococcal nuclease instead of ChIP, allowing for enhanced signal-to-noise ratio during sequencing.
- CUT&Tag sequencing, antibody-targeted controlled cleavage by transposase Tn5 instead of ChIP, allowing for enhanced signal-to-noise ratio during sequencing.
- Sono-Seq, identical to ChIP-Seq but skipping the immunoprecipitation step.
- HITS-CLIP[28][29] (also called CLIP-Seq), for finding interactions with RNA rather than DNA.
- PAR-CLIP, another method for identifying the binding sites of cellular RNA-binding proteins (RBPs).
- RIP-Chip, same goal and first steps, but does not use cross linking methods and uses microarray instead of sequencing
- SELEX, a method for finding a consensus binding sequence
- Competition-ChIP, to measure relative replacement dynamics on DNA.
- ChiRP-Seq to measure RNA-bound DNA and proteins.
- ChIP-exo uses exonuclease treatment to achieve up to single base-pair resolution
- ChIP-nexus improved version of ChIP-exo to achieve up to single base-pair resolution.
- DRIP-seq uses S9.6 antibody to precipitate three-stranded DND:RNA hybrids called R-loops.
- TCP-seq, principally similar method to measure mRNA translation dynamics.
- Calling Cards, uses a transposase to mark the sequence where a transcription factor binds.[30]
References
- ↑ Muhammad, Isiaka Ibrahim; Kong, Sze Ling; Akmar Abdullah, Siti Nor; Munusamy, Umaiyal (25 December 2019). "RNA-seq and ChIP-seq as Complementary Approaches for Comprehension of Plant Transcriptional Regulatory Mechanism". International Journal of Molecular Sciences. 21 (1): 167. doi:10.3390/ijms21010167. ISSN 1422-0067. PMC 6981605. PMID 31881735.
- ↑ Johnson DS, Mortazavi A, Myers RM, Wold B (June 2007). "Genome-wide mapping of in vivo protein-DNA interactions" (PDF). Science. 316 (5830): 1497–502. Bibcode:2007Sci...316.1497J. doi:10.1126/science.1141319. PMID 17540862. S2CID 519841.
- ↑ "Whole-Genome Chromatin IP Sequencing (ChIP-Seq)" (PDF). Illumina, Inc. 26 November 2007.
- ↑ Song, Lingyun; Crawford, Gregory E. (February 2010). "DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells". Cold Spring Harbor Protocols. 2010 (2): pdb.prot5384. doi:10.1101/pdb.prot5384. ISSN 1559-6095. PMC 3627383. PMID 20150147.
- ↑ Giresi, Paul G.; Kim, Jonghwan; McDaniell, Ryan M.; Iyer, Vishwanath R.; Lieb, Jason D. (June 2007). "FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin". Genome Research. 17 (6): 877–885. doi:10.1101/gr.5533506. ISSN 1088-9051. PMC 1891346. PMID 17179217.
- ↑ Kumar, Vibhor; Muratani, Masafumi; Rayan, Nirmala Arul; Kraus, Petra; Lufkin, Thomas; Ng, Huck Hui; Prabhakar, Shyam (July 2013). "Uniform, optimal signal processing of mapped deep-sequencing data". Nature Biotechnology. 31 (7): 615–622. doi:10.1038/nbt.2596. ISSN 1087-0156. PMID 23770639. S2CID 32510475.
- 1 2 3 4 "ChIP guide: epigenetics applications | Abcam". www.abcam.com. Retrieved 2 March 2020.
- ↑ Kim TH, Dekker J (April 2018). "Formaldehyde Cross-Linking". Cold Spring Harbor Protocols. 2018 (4): pdb.prot082594. doi:10.1101/pdb.prot082594. PMID 29610357.
- ↑ Kim TH, Dekker J (April 2018). "Preparation of Cross-Linked Chromatin for ChIP". Cold Spring Harbor Protocols. 2018 (4): pdb.prot082602. doi:10.1101/pdb.prot082602. PMID 29610358.
- ↑ Park, Peter J. (October 2009). "ChIP-seq: advantages and challenges of a maturing technology". Nature Reviews. Genetics. 10 (10): 669–680. doi:10.1038/nrg2641. ISSN 1471-0064. PMC 3191340. PMID 19736561.
- 1 2 Chen, Yiwen; Negre, Nicolas; Li, Qunhua; Mieczkowska, Joanna O.; Slattery, Matthew; Liu, Tao; Zhang, Yong; Kim, Tae-Kyung; He, Housheng Hansen; Zieba, Jennifer; Ruan, Yijun (June 2012). "Systematic evaluation of factors influencing ChIP-seq fidelity". Nature Methods. 9 (6): 609–614. doi:10.1038/nmeth.1985. ISSN 1548-7105. PMC 3477507. PMID 22522655.
- ↑ Jung, Youngsook L.; Luquette, Lovelace J.; Ho, Joshua W. K.; Ferrari, Francesco; Tolstorukov, Michael; Minoda, Aki; Issner, Robbyn; Epstein, Charles B.; Karpen, Gary H.; Kuroda, Mitzi I.; Park, Peter J. (May 2014). "Impact of sequencing depth in ChIP-seq experiments". Nucleic Acids Research. 42 (9): e74. doi:10.1093/nar/gku178. ISSN 1362-4962. PMC 4027199. PMID 24598259.
- ↑ Ho, Joshua W. K.; Bishop, Eric; Karchenko, Peter V.; Nègre, Nicolas; White, Kevin P.; Park, Peter J. (28 February 2011). "ChIP-chip versus ChIP-seq: lessons for experimental design and data analysis". BMC Genomics. 12: 134. doi:10.1186/1471-2164-12-134. ISSN 1471-2164. PMC 3053263. PMID 21356108.
- ↑ Jothi, et al. (2008). "Genome-wide identification of in vivo protein–DNA binding sites from ChIP-seq data". Nucleic Acids Res. 36 (16): 5221–5231. doi:10.1093/nar/gkn488. PMC 2532738. PMID 18684996.
- ↑ Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. (January 2005). "Genomic maps and comparative analysis of histone modifications in human and mouse". Cell. 120 (2): 169–81. doi:10.1016/j.cell.2005.01.001. PMID 15680324. S2CID 7193829.
- ↑ "HeLa S3 from ATCC | Biocompare.com". www.biocompare.com. Retrieved 21 March 2020.
- ↑ Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, et al. (August 2007). "Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing". Nature Methods. 4 (8): 651–7. doi:10.1038/nmeth1068. PMID 17558387. S2CID 28531263.
- ↑ Schmid CD, Bucher P (November 2007). "ChIP-Seq data reveal nucleosome architecture of human promoters". Cell. 131 (5): 831–2, author reply 832–3. doi:10.1016/j.cell.2007.11.017. PMID 18045524. S2CID 29234049.
- ↑ Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. (September 2010). "ChIP-Seq identification of weakly conserved heart enhancers". Nature Genetics. 42 (9): 806–10. doi:10.1038/ng.650. PMC 3138496. PMID 20729851.
- ↑ Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, et al. (February 2011). "Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans". Genome Research. 21 (2): 245–54. doi:10.1101/gr.114587.110. PMC 3032928. PMID 21177963.
- ↑ Kumar V, Muratani M, Rayan NA, Kraus P, Lufkin T, Ng HH, Prabhakar S (July 2013). "Uniform, optimal signal processing of mapped deep-sequencing data". Nature Biotechnology. 31 (7): 615–22. doi:10.1038/nbt.2596. PMID 23770639.
- ↑ Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. (2008). "Model-based analysis of ChIP-Seq (MACS)". Genome Biology. 9 (9): R137. doi:10.1186/gb-2008-9-9-r137. PMC 2592715. PMID 18798982.
- ↑ Xing H, Mo Y, Liao W, Zhang MQ (2012). "Genome-wide localization of protein-DNA binding and histone modification by a Bayesian change-point method with ChIP-seq data". PLOS Comput Biol. 8 (7): e1002613. Bibcode:2012PLSCB...8E2613X. doi:10.1371/journal.pcbi.1002613. PMC 5429005. PMID 22844240.
- ↑ Thomas R, Thomas S, Holloway AK, Pollard KS (2017). "Features that define the best ChIP-seq peak calling algorithms". Brief Bioinform. 18 (3): 441–450. doi:10.1093/bib/bbw035. PMC 5429005. PMID 27169896.
- ↑ Xu H, Wei CL, Lin F, Sung WK (October 2008). "An HMM approach to genome-wide identification of differential histone modification sites from ChIP-seq data". Bioinformatics. 24 (20): 2344–9. doi:10.1093/bioinformatics/btn402. PMID 18667444.
- ↑ Allhoff M, Seré K, Chauvistré H, Lin Q, Zenke M, Costa IG (December 2014). "Detecting differential peaks in ChIP-seq signals with ODIN". Bioinformatics. 30 (24): 3467–75. doi:10.1093/bioinformatics/btu722. PMID 25371479.
- 1 2 Xu, Jinrui; Kudron, Michelle M; Victorsen, Alec; Gao, Jiahao; Ammouri, Haneen N; Navarro, Fabio C P; Gevirtzman, Louis; Waterston, Robert H; White, Kevin P; Reinke, Valerie; Gerstein, Mark (21 December 2020). "To mock or not: a comprehensive comparison of mock IP and DNA input for ChIP-seq". Nucleic Acids Research. 49 (3): gkaa1155. doi:10.1093/nar/gkaa1155. ISSN 0305-1048. PMC 7897498. PMID 33347581.
- ↑ Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, et al. (November 2008). "HITS-CLIP yields genome-wide insights into brain alternative RNA processing". Nature. 456 (7221): 464–9. Bibcode:2008Natur.456..464L. doi:10.1038/nature07488. PMC 2597294. PMID 18978773.
- ↑ Darnell RB (2010). "HITS-CLIP: panoramic views of protein-RNA regulation in living cells". Wiley Interdisciplinary Reviews. RNA. 1 (2): 266–286. doi:10.1002/wrna.31. PMC 3222227. PMID 21935890.
- ↑ Wang H, Mayhew D, Chen X, Johnston M, Mitra RD (May 2011). "Calling Cards enable multiplexed identification of the genomic targets of DNA-binding proteins". Genome Research. 21 (5): 748–55. doi:10.1101/gr.114850.110. PMC 3083092. PMID 21471402.
External links
- ReMap catalogue: An integrative and uniform ChIP-Seq analysis of regulatory elements from +2800 ChIP-seq datasets, giving a catalogue of 80 million peaks from 485 transcription regulators.[1]
- ChIPBase database: a database for exploring transcription factor binding maps from ChIP-Seq data. It provides the most comprehensive ChIP-Seq data set for various cell/tissue types and conditions.
- GeneProf database and analysis tool: GeneProf is a freely accessible, easy-to-use analysis environment for ChIP-seq and RNA-seq data and comes with a large database of ready-analysed public experiments, e.g. for transcription factor binding and histone modifications.
- Differential Peak Calling Archived 15 May 2021 at the Wayback Machine: Tutorial for differential peak calling with ODIN.
- Bioinformatic analysis of ChIP-seq data: Comprehensive analysis of ChIP-seq data.[2]
- KLTepigenome: Uncovering correlated variability in epigenomic datasets using the Karhunen-Loeve transform.
- SignalSpider: a tool for probabilistic pattern discovery on multiple normalized ChIP-Seq signal profiles
- FullSignalRanker: a tool for regression and peak prediction on multiple normalized ChIP-Seq signal profiles
- ↑ Chèneby J, Gheorghe M, Artufel M, Mathelier A, Ballester B (January 2018). "ReMap 2018: an updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments". Nucleic Acids Research. 46 (D1): D267–D275. doi:10.1093/nar/gkx1092. PMC 5753247. PMID 29126285.
- ↑ Bailey T, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, et al. (2013). "Practical guidelines for the comprehensive analysis of ChIP-seq data". PLOS Computational Biology. 9 (11): e1003326. Bibcode:2013PLSCB...9E3326B. doi:10.1371/journal.pcbi.1003326. PMC 3828144. PMID 24244136.