An epigenetic clock is a biochemical test that can be used to measure age. The test is based on DNA methylation levels, measuring the accumulation of methyl groups to one's DNA molecules.
History
The strong effects of age on DNA methylation levels have been known since the late 1960s.[1] A vast literature describes sets of CpGs whose DNA methylation levels correlate with age.[2][3][4][5][6] The first robust demonstration that DNA methylation levels in saliva could generate age predictors with an average accuracy of 5.2 years was published by a UCLA team including Sven Bocklandt, Steve Horvath, and Eric Vilain in 2011 (Bocklandt et al. 2011).[7][8] The laboratories of Trey Ideker and Kang Zhang at the University of California, San Diego published the Hannum epigenetic clock (Hannum 2013),[9] which consisted of 71 markers that accurately estimate age based on blood methylation levels. The first multi-tissue epigenetic clock, Horvath's epigenetic clock, was developed by Steve Horvath, a professor of human genetics and biostatistics at UCLA (Horvath 2013).[10][11] Horvath spent over 4 years collecting publicly available Illumina DNA methylation data and identifying suitable statistical methods.[12]
The personal story behind the discovery was featured in Nature.[13] The age estimator was developed using 8,000 samples from 82 Illumina DNA methylation array datasets, encompassing 51 healthy tissues and cell types. The major innovation of Horvath's epigenetic clock lies in its wide applicability: the same set of 353 CpGs and the same prediction algorithm is used irrespective of the DNA source within the organism, i.e. it does not require any adjustments or offsets.[10] This property allows one to compare the ages of different areas of the human body using the same aging clock. Shortly afterwards, a derivation of Horvath's clock, the IEAA (Intrinsic Epigenetic Age Acceleration), an estimator based on the cellular composition of the blood, was developed.
A second generation of epigenetic clocks emerged a few years later and improved on the first in age estimation. This was thanks to the incorporation not only of epigenetic variants such as DNA methylation but also environmental variants such as smoking or chronological age. Among these clocks, the PhenoAge and GrimAge clocks stand out. PhenoAge is an epigenetic clock that takes chronological age into account, and GrimAge uses the mortality risks of age together with the smoking variant among others as a risk factor. Taking into account environmental variants allows GrimAge to outperform any other epigenetic clock in "predicting death".
Third-generation epigenetic clocks are designed to be applicable across multiple species simultaneously. Specifically, pan-mammalian epigenetic clocks determine the age of tissues from all mammalian species by analyzing cytosine methylation in DNA regions that are highly conserved.[14]
New age estimation tools have been developed continuously, which also facilitate the prognosis of certain diseases.
Relationship to a cause of biological aging
It is not yet known what exactly is measured by DNA methylation age. Horvath hypothesized that DNA methylation age measures the cumulative effect of an epigenetic maintenance system but details are unknown. The fact that DNA methylation age of blood predicts all-cause mortality in later life[15][16][17][18] has been used to argue that it relates to a process that causes aging.[15] However, if a particular CpG played a direct causal role in the aging process, the mortality it created would make it less likely to be observed in older individuals, making the site less likely to have been chosen as a predictor; the 353 clock CpGs therefore likely have no causal effect whatsoever.[19] Rather, the epigenetic clock captures an emergent property of the epigenome.
Epigenetic clock theory of aging
In 2010, a new unifying model of aging and the development of complex diseases was proposed, incorporating classical aging theories and epigenetics.[20][21] Horvath and Raj[22] extended this theory, proposing an epigenetic clock theory of aging with the following tenets:
- Biological aging results as an unintended consequence of both developmental programs and maintenance program, the molecular footprints of which give rise to DNA methylation age estimators.
- The precise mechanisms linking the innate molecular processes (underlying DNAm age) to the decline in tissue function probably relate to both intracellular changes (leading to a loss of cellular identity) and subtle changes in cell composition, for example, fully functioning somatic stem cells.
- At the molecular level, DNAm age is a proximal readout of a collection of innate aging processes that conspire with other, independent root causes of aging to the detriment of tissue function.
Motivation for biological clocks
In general, biological aging clocks and biomarkers of aging are expected to find many uses in biological research since age is a fundamental characteristic of most organisms. Accurate measures of biological age (biological aging clocks) could be useful for
- testing the validity of various theories of biological aging,
- diagnosing various age related diseases and for defining cancer subtypes,
- predicting/prognosticating the onset of various diseases,
- serving as surrogate markers for evaluating therapeutic interventions including rejuvenation approaches,
- studying developmental biology and cell differentiation,
- forensic applications, for example to estimate the age of a suspect based on blood left on a crime scene.
Overall, biological clocks are expected to be useful for studying what causes aging and what can be done against it. However, they can only capture the effects of interventions that affect the rate of future aging, i.e. the slope of the Gompertz curve by which mortality increases with age, and not that of interventions that act at one moment in time, e.g. to lower mortality across all ages, i.e. the intercept of the Gompertz curve.[19]
Properties of Horvath's clock
The clock is defined as an age estimation method based on 353 epigenetic markers on the DNA. The 353 markers measure DNA methylation of CpG dinucleotides. Estimated age ("predicted age" in mathematical usage), also referred to as DNA methylation age, has the following properties: first, it is close to zero for embryonic and induced pluripotent stem cells; second, it correlates with cell passage number; third, it gives rise to a highly heritable measure of age acceleration; and, fourth, it is applicable to chimpanzee tissues (which are used as human analogs for biological testing purposes). Organismal growth (and concomitant cell division) leads to a high ticking rate of the epigenetic clock that slows down to a constant ticking rate (linear dependence) after adulthood (age 20).[10] The fact that DNA methylation age of blood predicts all-cause mortality in later life even after adjusting for known risk factors[15][16] is compatible with a variety of causal relationships, e.g. a common cause for both. Similarly, markers of physical and mental fitness are associated with the epigenetic clock (lower abilities associated with age acceleration).[23] It systematically underestimates age from older individuals.[24]
Salient features of Horvath's epigenetic clock include its applicability to a broad spectrum of tissues and cell types. Since it allows one to contrast the ages of different tissues from the same subject, it can be used to identify tissues that show evidence of accelerated age due to disease.
Genetic estimators in the Horvath clock
The Horvath clock, specifically the IEAA variant, is associated with several ageing-related genes:14
- TRIM59: of the tripartite motif family, strongly associated with chronological age and whose expression has been observed in multiple cancers
- SMC4: inhibits cellular senescence, an established hallmark of ageing
- KPNA4: member of the importin family, nuclear transport receptors. Dysfunction of nuclear transport has been proposed as a marker of ageing
- CD46: encodes a regulator of T-cell function and the complement system, a key component of the innate immune system where it promotes inflammation
- ATP8B4: encodes for a lipid transporter protein and contains variants that have been reported in association with Alzheimer's disease
- CXXC4: encodes Idax, an inhibitor of Wnt signalling[25]
Statistical approach
The basic approach is to form a weighted average of the 353 clock CpGs, which is then transformed to DNAm age using a calibration function. The calibration function reveals that the epigenetic clock has a high ticking rate until adulthood, after which it slows to a constant ticking rate. Using the training data sets, Horvath used a penalized regression model (Elastic net regularization) to regress a calibrated version of chronological age on 21,369 CpG probes that were present both on the Illumina 450K and 27K platform and had fewer than 10 missing values. DNAm age is defined as estimated ("predicted") age. The elastic net predictor automatically selected 353 CpGs. 193 of the 353 CpGs correlate positively with age while the remaining 160 CpGs correlate negatively with age. R software and a freely available web-based tool can be found at the following webpage.[26]
Accuracy
The median error of estimated age is 3.6 years across a wide spectrum of tissues and cell types,[10] although this increases for older individuals[24] The epigenetic clock performs well in heterogeneous tissues (for example, whole blood, peripheral blood mononuclear cells, cerebellar samples, occipital cortex, buccal epithelium, colon, adipose, kidney, liver, lung, saliva, uterine cervix, epidermis, muscle) as well as in individual cell types such as CD4 T cells, CD14 monocytes, glial cells, neurons, immortalized B cells, mesenchymal stromal cells.[10] However, accuracy depends to some extent on the source of the DNA.
Comparison with other biological clocks
The epigenetic clock leads to a chronological age prediction that has a Pearson correlation coefficient of r = 0.96 with chronological age (Figure 2 in[10]). Thus the age correlation is close to its maximum possible correlation value of 1. Other biological clocks are based on a) telomere length, b) p16INK4a expression levels (also known as INK4a/ARF locus),[27] and c) microsatellite mutations.[28] The correlation between chronological age and telomere length is r = −0.51 in women and r = −0.55 in men.[29] The correlation between chronological age and expression levels of p16INK4a in T cells is r = 0.56.[30]
Applications of Horvath's clock
By contrasting DNA methylation age (estimated age) with chronological age, one can define measures of age acceleration. Age acceleration can be defined as the difference between DNA methylation age and chronological age. Alternatively, it can be defined as the residual that results from regressing DNAm age on chronological age. The latter measure is attractive because it does not correlate with chronological age. A positive/negative value of epigenetic age acceleration suggests that the underlying tissue ages faster/slower than expected.
Genetic studies of epigenetic age acceleration
The broad sense heritability (defined via Falconer's formula) of age acceleration of blood from older subjects is around 40% but it appears to be much higher in newborns.[10] Similarly, the age acceleration of brain tissue (prefrontal cortex) was found to be 41% in older subjects.[31] Genome-wide association studies (GWAS) of epigenetic age acceleration in postmortem brain samples have identified several SNPs at a genomewide significance level.[32][33] GWAS of age acceleration in blood have identified several genome-wide significant genetic loci including the telomerase reverse transcriptase gene (TERT) locus.[34] Genetic variants associated with longer leukocyte telomere length in TERT gene paradoxically confer higher epigenetic age acceleration in blood.[34]
Lifestyle factors
In general, lifestyle factors have only weak associations with epigenetic age acceleration in blood.[35][36][37] Cross sectional studies of extrinsic epigenetic aging rates in blood show reduced epigenetic aging correlates with higher education, eating a high plant diet with lean meats, moderate alcohol consumption, and physical activity[36] and the risks associated with metabolic syndrome. However, studies suggest that high levels of alcohol consumption are associated with accelerated aging of certain epigenetic clocks.[37]
Obesity and metabolic syndrome
The epigenetic clock was used to study the relationship between high body mass index (BMI) and the DNA methylation ages of human blood, liver, muscle and adipose tissue.[38] A significant correlation (r = 0.42) between BMI and epigenetic age acceleration could be observed for the liver. A much larger sample size (n = 4200 blood samples) revealed a weak but statistically significant correlation (r = 0.09) between BMI and intrinsic age acceleration of blood.[35] The same large study found that various biomarkers of metabolic syndrome (glucose-, insulin-, triglyceride levels, C-reactive protein, waist-to-hip ratio) were associated with epigenetic age acceleration in blood.[35] Conversely, high levels of HDL cholesterol were associated with a lower epigenetic aging rate of blood.[35] Other research suggests very strong associations between higher body mass index, waist-to-hip ratio, and waist circumference and accelerated epigenetic clocks, with evidence that physical activity may lessen these effects.[36]
Female breast tissue is older than expected
DNAm age is higher than chronological age in female breast tissue that is adjacent to breast cancer tissue.[10] Since normal tissue which is adjacent to other cancer types does not exhibit a similar age acceleration effect, this finding suggests that normal female breast tissue ages faster than other parts of the body.[10] Similarly, normal breast tissue samples from women without cancer have been found to be substantially older than blood samples collected from the same women at the same time.[39]
Female breast cancer
In a study of three epigenetic clocks and breast cancer risk, DNAm age was found to be accelerated in blood samples of cancer-free women, years before diagnosis.[40]
Cancer tissue
Cancer tissues show both positive and negative age acceleration effects. For most tumor types, no significant relationship can be observed between age acceleration and tumor morphology (grade/stage).[10][41] On average, cancer tissues with mutated TP53 have a lower age acceleration than those without it.[10] Further, cancer tissues with high age acceleration tend to have fewer somatic mutations than those with low age acceleration.[10][41] Age acceleration is highly related to various genomic aberrations in cancer tissues. Somatic mutations in estrogen receptors or progesterone receptors are associated with accelerated DNAm age in breast cancer.[10] Colorectal cancer samples with a BRAF (V600E) mutation or promoter hypermethylation of the mismatch repair gene MLH1 are associated with an increased age acceleration.[10] Age acceleration in glioblastoma multiforme samples is highly significantly associated with certain mutations in H3F3A.[10] One study suggests that the epigenetic age of blood tissue may be prognostic of lung cancer incidence.[42]
Trisomy 21 (Down syndrome)
Down syndrome entails an increased risk of many chronic diseases that are typically associated with older age. The clinical manifestations of accelerated aging suggest that trisomy 21 increases the biological age of tissues, but molecular evidence for this hypothesis has been sparse. According to the epigenetic clock, trisomy 21 significantly increases the age of blood and brain tissue (on average by 6.6 years).[43]
Alzheimer's disease related neuropathology
Epigenetic age acceleration of the human prefrontal cortex was found to be correlated with several neuropathological measurements that play a role in Alzheimer's disease[31] Further, it was found to be associated with a decline in global cognitive functioning, and memory functioning among individuals with Alzheimer's disease.[31] The epigenetic age of blood relates to cognitive functioning in the elderly.[23] Overall, these results strongly suggest that the epigenetic clock lends itself for measuring the biological age of the brain.
Cerebellum ages slowly
It has been difficult to identify tissues that seem to evade aging due to the lack of biomarkers of tissue age that allow one to contrast compare the ages of different tissues. An application of epigenetic clock to 30 anatomic sites from six centenarians and younger subjects revealed that the cerebellum ages slowly: it is about 15 years younger than expected in a centenarian.[44] This finding might explain why the cerebellum exhibits fewer neuropathological hallmarks of age related dementias compared to other brain regions. In younger subjects (e.g. younger than 70), brain regions and brain cells appear to have roughly the same age.[10][44] Several SNPs and genes have been identified that relate to the epigenetic age of the cerebellum.[32]
Huntington's disease
Huntington's disease has been found to increase the epigenetic aging rates of several human brain regions.[45]
Centenarians age slowly
The offspring of semi-supercentenarians (subjects who reached an age of 105–109 years) have a lower epigenetic age than age-matched controls (age difference = 5.1 years in blood) and centenarians are younger (8.6 years) than expected based on their chronological age.[18]
HIV infection
Infection with the Human Immunodeficiency Virus-1 (HIV) is associated with clinical symptoms of accelerated aging, as evidenced by increased incidence and diversity of age-related illnesses at relatively young ages. But it has been difficult to detect an accelerated aging effect on a molecular level. An epigenetic clock analysis of human DNA from HIV+ subjects and controls detected a significant age acceleration effect in brain (7.4 years) and blood (5.2 years) tissue due to HIV-1 infection.[46] These results are consistent with an independent study that also found an age advancement of 5 years in blood of HIV patients and a strong effect of the HLA locus.[47]
Parkinson's disease
A large-scale study suggests that the blood of Parkinson's disease subjects, in particular, their granulocyte ratio, exhibits (relatively weak) accelerated aging effects.[48]
Developmental disorder: syndrome X
Children with a very rare disorder known as syndrome X maintain the façade of persistent toddler-like features while aging from birth to adulthood. Since the physical development of these children is dramatically delayed, these children appear to be a toddler or at best a preschooler. According to an epigenetic clock analysis, blood tissue from syndrome X cases is not younger than expected.[49]
Menopause accelerates epigenetic aging
The following results strongly suggest that the loss of female hormones resulting from menopause accelerates the epigenetic aging rate of blood and possibly that of other tissues.[50] First, early menopause has been found to be associated with an increased epigenetic age acceleration of blood.[50] Second, surgical menopause (due to bilateral oophorectomy) is associated with epigenetic age acceleration in blood and saliva. Third, menopausal hormone therapy, which mitigates hormonal loss, is associated with a negative age acceleration of buccal cells (but not of blood cells).[50] Fourth, genetic markers that are associated with early menopause are also associated with increased epigenetic age acceleration in blood.[50]
Cellular senescence versus epigenetic aging
A confounding aspect of biological aging is the nature and role of senescent cells. It is unclear whether the three major types of cellular senescence, namely replicative senescence, oncogene-induced senescence and DNA damage-induced senescence are descriptions of the same phenomenon instigated by different sources, or if each of these is distinct, and how they are associated with epigenetic aging. Induction of replicative senescence (RS) and oncogene-induced senescence (OIS) were found to be accompanied by epigenetic aging of primary cells but senescence induced by DNA damage was not, even though RS and OIS activate the cellular DNA damage response pathway.[51] These results highlight the independence of cellular senescence from epigenetic aging. Consistent with this, telomerase-immortalised cells continued to age (according to the epigenetic clock) without having been treated with any senescence inducers or DNA-damaging agents, re-affirming the independence of the process of epigenetic ageing from telomeres, cellular senescence, and the DNA damage response pathway. Although the uncoupling of senescence from cellular aging appears at first sight to be inconsistent with the fact that senescent cells contribute to the physical manifestation of organism ageing, as demonstrated by Baker et al., where removal of senescent cells slowed down aging.[52]
The epigenetic clock analysis of senescence, however, suggests that cellular senescence is a state that cells are forced into as a result of external pressures such as DNA damage, ectopic oncogene expression and exhaustive proliferation of cells to replenish those eliminated by external/environmental factors.[51] These senescent cells, in sufficient numbers, will probably cause the deterioration of tissues, which is interpreted as organism ageing. However, at the cellular level, aging, as measured by the epigenetic clock, is distinct from senescence. It is an intrinsic mechanism that exists from the birth of the cell and continues. This implies that if cells are not shunted into senescence by the external pressures described above, they would still continue to age. This is consistent with the fact that mice with naturally long telomeres still age and eventually die even though their telomere lengths are far longer than the critical limit, and they age prematurely when their telomeres are forcibly shortened, due to replicative senescence. Therefore, cellular senescence is a route by which cells exit prematurely from the natural course of cellular aging.[51]
Effect of sex and race/ethnicity
Men age faster than women according to epigenetic age acceleration in blood, brain, saliva, but it depends on the structure being researched and the lifestyle.[53] The epigenetic clock method applies to all examined racial/ethnic groups in the sense that DNAm age is highly correlated with chronological age. But ethnicity can be associated with epigenetic age acceleration.[53] For example, the blood of Hispanics and the Tsimané ages more slowly than that of other populations which might explain the Hispanic mortality paradox.[53]
Rejuvenation effect due to stem cell transplantation in blood
Hematopoietic stem cell transplantation, which transplants these cells from a young donor to an older recipient, rejuvenates the epigenetic age of blood to that of the donor. However, graft-versus-host disease is associated with increased DNA methylation age.[54]
Progeria
Adult progeria also known as Werner syndrome is associated with epigenetic age acceleration in blood.[55] Fibroblast samples from children with Hutchinson-Gilford Progeria exhibit accelerated epigenetic aging effects according to the "skin & blood" epigenetic clock but not according to the original pan tissue clock from Horvath.[56]
Biological mechanism behind the epigenetic clock
Despite the fact that biomarkers of ageing based on DNA methylation data have enabled accurate age estimates for any tissue across the entire life course, the precise biological mechanism behind the epigenetic clock is currently unknown.[22] However, epigenetic biomarkers may help to address long-standing questions in many fields, including the central question: why do we age? To understand the essence of mechanisms behind the epigenetic clock, it would be advisable to make a comparison and find the relationship between the readings of the epigenetic clock and the transcriptome aging clock.[57] The following explanations have been proposed for now in the literature.
Possible explanation 1: Epigenomic maintenance system
Horvath hypothesized that his clock arises from a methylation footprint left by an epigenomic maintenance system.[10]
Possible explanation 2: Unrepaired DNA damages
Endogenous DNA damages occur frequently including about 50 double-strand DNA breaks per cell cycle[58] and about 10,000 oxidative damages per day (see DNA damage (naturally occurring)). During repair of double-strand breaks many epigenetic alterations are introduced, and in a percentage of cases epigenetic alterations remain after repair is completed, including increased methylation of CpG island promoters.[59][60][61] Similar, but usually transient epigenetic alterations were recently found during repair of oxidative damages caused by H2O2, and it was suggested that occasionally these epigenetic alterations may also remain after repair.[62] These accumulated epigenetic alterations may contribute to the epigenetic clock. Accumulation of epigenetic alterations may parallel the accumulation of un-repaired DNA damages that are proposed to cause aging (see DNA damage theory of aging).
Other age estimators based on DNA methylation levels
Several other age estimators have been described in the literature.
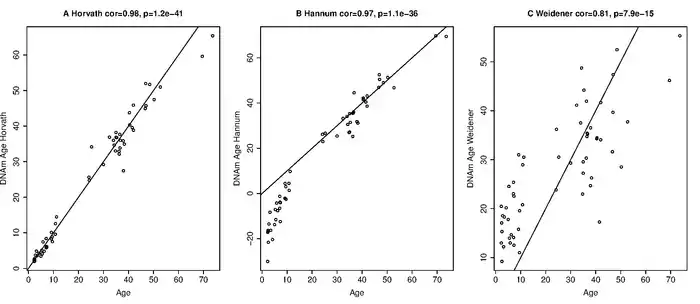
1) Weidner et al. (2014) describe an age estimator for DNA from blood that uses only three CpG sites of genes hardly affected by aging (cg25809905 in integrin, alpha 2b (ITGA2B); cg02228185 in aspartoacylase (ASPA) and cg17861230 in phosphodiesterase 4C, cAMP specific (PDE4C)).[63] The age estimator by Weidener et al. (2014) applies only to blood. Even in blood this sparse estimator is far less accurate than Horvath's epigenetic clock (Horvath 2014) when applied to data generated by the Illumina 27K or 450K platforms.[64] But the sparse estimator was developed for pyrosequencing data and is highly cost effective.[65]
2) Hannum et al. (2013)[9] report several age estimators: one for each tissue type. Each of these estimators requires covariate information (e.g. gender, body mass index, batch). The authors mention that each tissue led to a clear linear offset (intercept and slope). Therefore, the authors had to adjust the blood-based age estimator for each tissue type using a linear model. When the Hannum estimator is applied to other tissues, it leads to a high error (due to poor calibration) as can be seen from Figure 4A in Hannum et al. (2013). Hannum et al. adjusted their blood-based age estimator (by adjusting the slope and the intercept term) in order to apply it to other tissue types. Since this adjustment step removes differences between tissue, the blood-based estimator from Hannum et al. cannot be used to compare the ages of different tissues/organs. In contrast, a salient characteristic of the epigenetic clock is that one does not have to carry out such a calibration step:[10] it always uses the same CpGs and the same coefficient values. Therefore, Horvath's epigenetic clock can be used to compare the ages of different tissues/cells/organs from the same individual. While the age estimators from Hannum et al. cannot be used to compare the ages of different normal tissues, they can be used to compare the age of a cancerous tissue with that of a corresponding normal (non-cancerous) tissue. Hannum et al. reported pronounced age acceleration effects in all cancers. In contrast, Horvath's epigenetic clock[41][66] reveals that some cancer types (e.g. triple negative breast cancers or uterine corpus endometrial carcinoma) exhibit negative age acceleration, i.e. cancer tissue can be much younger than expected. An important difference relates to additional covariates. Hannum's age estimators make use of covariates such as gender, body mass index, diabetes status, ethnicity, and batch. Since new data involve different batches, one cannot apply it directly to new data. However, the authors present coefficient values for their CpGs in Supplementary Tables which can be used to define an aggregate measure that tends to be strongly correlated with chronological age but may be poorly calibrated (i.e. lead to high errors).
3) Giuliani et al. identify genomic regions whose DNA methylation level correlates with age in human teeth. They propose the evaluation of DNA methylation at ELOVL2, FHL2, and PENK genes in DNA recovered from both cementum and pulp of the same modern teeth.[67] They wish to apply this method also to historical and relatively ancient human teeth.
4) Galkin et al. used deep neural networks to train an epigenetic aging clock of unprecedented accuracy using >6,000 blood samples.[68] The clock uses information from 1000 CpG sites and predicts people with certain conditions older than healthy controls: IBD, frontotemporal dementia, ovarian cancer, obesity. The aging clock is planned to be released for public use in 2021 by an Insilico Medicine spinoff company Deep Longevity.
In a multicenter benchmarking study 18 research groups from three continents compared all promising methods for analyzing DNA methylation in the clinic and identified the most accurate methods, having concluded that epigenetic tests based on DNA methylation are a mature technology ready for broad clinical use.[69]
5) McCartney et al. Plasminogen activator inhibitor 1 (PAI1) can also be used as an age estimator related to DNA methylation levels, as it has been shown to exhibit stronger associations with cardiometabolic disease than some epigenetic clocks.
6) de Lima Camillo et al. (2022)[70] used an optimized deep neural network to create a highly accurate, robust pan-tissue epigenetic clock. The predictor, dubbed AltumAge, was trained on 142 datasets and uses 20,318 CpG sites to achieve one of the lowest reported median absolute errors for human epigenetic age prediction of 2.153 years. Partly, that increase in performance of Horvath's linear method is due to AltumAge's ability to detect CpG-CpG interactions. AltumAge predicts higher age for people with autism, HIV, multiple sclerosis, non-alcoholic fatty liver disease, type 2 diabetes, and atherosclerosis. In contrast to Horvath's clock, AltumAge predicts cancer to be older than normal tissues. The code for the clock is publicly available.[71]
Other species
Wang et al., (in mice livers)[72] and Petkovich et al. (based on mice blood DNA methylation profiles)[73] examined whether mice and humans experience similar patterns of change in the methylome with age. They found that mice treated with lifespan-extending interventions (such as calorie restriction or dietary rapamycin) were significantly younger in epigenetic age than their untreated, wild-type age-matched controls. Mice age predictors also detects the longevity effects of gene knockouts, and rejuvenation of fibroblast-derived iPSCs.
Mice multi-tissue age predictor based on DNA methylation at 329 unique CpG sites reached a median absolute error of less than four weeks (~5 percent of lifespan). An attempt to use the human clock sites in mice for age predictions showed that the human clock is not fully conserved in mice.[74] Differences between human and mouse clocks suggests that epigenetic clocks need to be trained specifically for different species.[75]
A novel method of ageing lobsters was published in 2021 that used a ribosomal DNA methylation-based clock which may allow non-invasive sampling and ageing of wild European lobster populations (Homarus gammarus).[76]
Changes to DNA methylation patterns have great potential for age estimation and biomarker search in domestic and wild animals.[77]
The Mammalian Methylation Consortium generated DNA methylation data in highly conserved stretches of DNA.[78] Using these data, epigenetic clocks have been published for hundreds of different mammalian species. In particular, pan-mammalian clocks apply to tissues from all mammalian species.[14]
References
- ↑ Berdyshev GD, Korotaev GK, Boiarskikh GV, Vaniushin BF (1967). "[Nucleotide composition of DNA and RNA from somatic tissues of humpback and its changes during spawning]". Biokhimiia. 32 (5): 988–993. PMID 5628601.
- ↑ Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, et al. (April 2010). "Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains". Genome Research. 20 (4): 434–439. doi:10.1101/gr.103101.109. PMC 2847746. PMID 20219945.
- ↑ Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, et al. (April 2010). "Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer". Genome Research. 20 (4): 440–446. doi:10.1101/gr.103606.109. PMC 2847747. PMID 20219944.
- ↑ Koch CM, Wagner W (October 2011). "Epigenetic-aging-signature to determine age in different tissues". Aging. 3 (10): 1018–1027. doi:10.18632/aging.100395. PMC 3229965. PMID 22067257.
- ↑ Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, et al. (October 2012). "Aging effects on DNA methylation modules in human brain and blood tissue". Genome Biology. 13 (10): R97. doi:10.1186/gb-2012-13-10-r97. PMC 4053733. PMID 23034122.
- ↑ Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, et al. (2012). "Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population". PLOS Genetics. 8 (4): e1002629. doi:10.1371/journal.pgen.1002629. PMC 3330116. PMID 22532803.
- ↑ "Scientists discover new biological clock with age-measuring potential". Forbes. 21 October 2013. Retrieved 21 October 2013.
- ↑ Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, Vilain E (2011). "Epigenetic predictor of age". PLOS ONE. 6 (6): e14821. Bibcode:2011PLoSO...614821B. doi:10.1371/journal.pone.0014821. PMC 3120753. PMID 21731603.
- 1 2 3 Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. (January 2013). "Genome-wide methylation profiles reveal quantitative views of human aging rates". Molecular Cell. 49 (2): 359–367. doi:10.1016/j.molcel.2012.10.016. PMC 3780611. PMID 23177740.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 Horvath S (2013). "DNA methylation age of human tissues and cell types". Genome Biology. 14 (10): R115. doi:10.1186/gb-2013-14-10-r115. PMC 4015143. PMID 24138928.
- ↑ "Scientist uncovers internal clock able to measure age of most human tissues; Women's breast tissue ages faster than rest of body". ScienceDaily. 20 October 2013. Retrieved 22 October 2013.
- ↑ "Novel epigenetic clock predicts tissue age". Biome. October 21, 2013. Archived from the original on December 31, 2013.
- ↑ Gibbs WW (April 2014). "Biomarkers and ageing: The clock-watcher". Nature. 508 (7495): 168–170. Bibcode:2014Natur.508..168G. doi:10.1038/508168a. PMID 24717494.
- 1 2 Lu AT, Fei Z, Haghani A, et al. (2023). "Universal DNA methylation age across mammalian tissues [published correction appears in Nat Aging. 2023 Sep 6;]". Nat Aging. 3 (9): 1144–1166. doi:10.1038/s43587-023-00462-6. hdl:10023/28280.
- 1 2 3 Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai PC, et al. (September 2016). "DNA methylation-based measures of biological age: meta-analysis predicting time to death". Aging. 8 (9): 1844–1865. doi:10.18632/aging.101020. PMC 5076441. PMID 27690265.
- 1 2 Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, et al. (January 2015). "DNA methylation age of blood predicts all-cause mortality in later life". Genome Biology. 16 (1): 25. doi:10.1186/s13059-015-0584-6. PMC 4350614. PMID 25633388.
- ↑ Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, Christensen K (February 2016). "DNA methylation age is associated with mortality in a longitudinal Danish twin study". Aging Cell. 15 (1): 149–154. doi:10.1111/acel.12421. PMC 4717264. PMID 26594032.
- 1 2 Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, et al. (December 2015). "Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring". Aging. 7 (12): 1159–1170. doi:10.18632/aging.100861. PMC 4712339. PMID 26678252.
- 1 2 Nelson PG, Promislow DE, Masel J (February 2020). "Biomarkers for Aging Identified in Cross-sectional Studies Tend to Be Non-causative". The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 75 (3): 466–472. doi:10.1093/gerona/glz174. PMC 7457180. PMID 31353411.
- ↑ Schumacher, Axel (2010). Trygve Tollefsbol, editors, Handbook of Epigenetics: The New Molecular and Medical Genetics. Elsevier. pp. 405–422. ISBN 978-0123757098.
- ↑ Schumacher, Axel (21 August 2017). Trygve Tollefsbol, editors, Handbook of Epigenetics: The New Molecular and Medical Genetics - 2nd Edition. Elsevier. p. Ch. 26. ISBN 9780128053881.
- 1 2 Horvath S, Raj K (June 2018). "DNA methylation-based biomarkers and the epigenetic clock theory of ageing". Nature Reviews. Genetics. 19 (6): 371–384. doi:10.1038/s41576-018-0004-3. PMID 29643443. S2CID 4709691.
- 1 2 Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, et al. (August 2015). "The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936". International Journal of Epidemiology. 44 (4): 1388–1396. doi:10.1093/ije/dyu277. PMC 4588858. PMID 25617346.
- 1 2 El Khoury LY, Gorrie-Stone T, Smart M, Hughes A, Bao Y, Andrayas A, et al. (December 2019). "Systematic underestimation of the epigenetic clock and age acceleration in older subjects". Genome Biology. 20 (1): 283. doi:10.1186/s13059-019-1810-4. PMC 6915902. PMID 31847916.
- ↑ McCartney DL, Min JL, Richmond RC, Lu AT, Sobczyk MK, Davies G, et al. (June 2021). "Genome-wide association studies identify 137 genetic loci for DNA methylation biomarkers of aging". Genome Biology. 22 (1): 194. doi:10.1186/s13059-021-02398-9. PMC 8243879. PMID 34187551.
 Text was copied from this source, which is available under a Creative Commons Attribution 4.0 International License.
Text was copied from this source, which is available under a Creative Commons Attribution 4.0 International License. - ↑ DNA methylation age calculator
- ↑ Collado M, Blasco MA, Serrano M (July 2007). "Cellular senescence in cancer and aging". Cell. 130 (2): 223–233. doi:10.1016/j.cell.2007.07.003. PMID 17662938. S2CID 18689141.
- ↑ Forster P, Hohoff C, Dunkelmann B, Schürenkamp M, Pfeiffer H, Neuhuber F, Brinkmann B (March 2015). "Elevated germline mutation rate in teenage fathers". Proceedings. Biological Sciences. 282 (1803): 20142898. doi:10.1098/rspb.2014.2898. PMC 4345458. PMID 25694621.
- ↑ Nordfjäll K, Svenson U, Norrback KF, Adolfsson R, Roos G (March 2010). "Large-scale parent-child comparison confirms a strong paternal influence on telomere length". European Journal of Human Genetics. 18 (3): 385–389. doi:10.1038/ejhg.2009.178. PMC 2987222. PMID 19826452.
- ↑ Wang Y, Zang X, Wang Y, Chen P (2012). "High expression of p16INK4a and low expression of Bmi1 are associated with endothelial cellular senescence in the human cornea". Molecular Vision. 18: 803–815. PMC 3324359. PMID 22509111.
- 1 2 3 Levine ME, Lu AT, Bennett DA, Horvath S (December 2015). "Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer's disease related cognitive functioning". Aging. 7 (12): 1198–1211. doi:10.18632/aging.100864. PMC 4712342. PMID 26684672.
- 1 2 Lu AT, Hannon E, Levine ME, Hao K, Crimmins EM, Lunnon K, et al. (February 2016). "Genetic variants near MLST8 and DHX57 affect the epigenetic age of the cerebellum". Nature Communications. 7: 10561. Bibcode:2016NatCo...710561L. doi:10.1038/ncomms10561. PMC 4740877. PMID 26830004.
- ↑ Lu AT, Hannon E, Levine ME, Crimmins EM, Lunnon K, Mill J, et al. (May 2017). "Genetic architecture of epigenetic and neuronal ageing rates in human brain regions". Nature Communications. 8 (15353): 15353. Bibcode:2017NatCo...815353L. doi:10.1038/ncomms15353. PMC 5454371. PMID 28516910.
- 1 2 Lu AT, Xue L, Salfati EL, Chen BH, Ferrucci L, Levy D, et al. (January 2018). "GWAS of epigenetic aging rates in blood reveals a critical role for TERT". Nature Communications. 9 (1): 387. Bibcode:2018NatCo...9..387L. doi:10.1038/s41467-017-02697-5. PMC 5786029. PMID 29374233.
- 1 2 3 4 Quach A, Levine ME, Tanaka T, Lu AT, Chen BH, Ferrucci L, et al. (February 2017). "Epigenetic clock analysis of diet, exercise, education, and lifestyle factors". Aging. 9 (2): 419–446. doi:10.18632/aging.101168. PMC 5361673. PMID 28198702.
- 1 2 3 Kresovich JK, Garval EL, Martinez Lopez AM, Xu Z, Niehoff NM, White AJ, et al. (June 2021). "Associations of Body Composition and Physical Activity Level With Multiple Measures of Epigenetic Age Acceleration". American Journal of Epidemiology. 190 (6): 984–993. doi:10.1093/aje/kwaa251. PMC 8168202. PMID 33693587.
- 1 2 Kresovich JK, Martinez Lopez AM, Garval EL, Xu Z, White AJ, Sandler DP, Taylor JA (November 2021). "Alcohol Consumption and Methylation-Based Measures of Biological Age". The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 76 (12): 2107–2111. doi:10.1093/gerona/glab149. PMC 8599006. PMID 34038541.
- ↑ Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schönfels W, Ahrens M, et al. (October 2014). "Obesity accelerates epigenetic aging of human liver". Proceedings of the National Academy of Sciences of the United States of America. 111 (43): 15538–15543. Bibcode:2014PNAS..11115538H. doi:10.1073/pnas.1412759111. PMC 4217403. PMID 25313081.
- ↑ Sehl ME, Henry JE, Storniolo AM, Ganz PA, Horvath S (July 2017). "DNA methylation age is elevated in breast tissue of healthy women". Breast Cancer Research and Treatment. 164 (1): 209–219. doi:10.1007/s10549-017-4218-4. PMC 5487725. PMID 28364215.
- ↑ Kresovich JK, Xu Z, O'Brien KM, Weinberg CR, Sandler DP, Taylor JA (October 2019). "Methylation-Based Biological Age and Breast Cancer Risk". Journal of the National Cancer Institute. 111 (10): 1051–1058. doi:10.1093/jnci/djz020. PMC 6792078. PMID 30794318.
- 1 2 3 Horvath S (May 2015). "Erratum to: DNA methylation age of human tissues and cell types". Genome Biology. 16 (1): 96. doi:10.1186/s13059-015-0649-6. PMC 4427927. PMID 25968125.
- ↑ Levine ME, Hosgood HD, Chen B, Absher D, Assimes T, Horvath S (September 2015). "DNA methylation age of blood predicts future onset of lung cancer in the women's health initiative". Aging. 7 (9): 690–700. doi:10.18632/aging.100809. PMC 4600626. PMID 26411804.
- ↑ Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, et al. (June 2015). "Accelerated epigenetic aging in Down syndrome". Aging Cell. 14 (3): 491–495. doi:10.1111/acel.12325. PMC 4406678. PMID 25678027.
- 1 2 Horvath S, Mah V, Lu AT, Woo JS, Choi OW, Jasinska AJ, et al. (May 2015). "The cerebellum ages slowly according to the epigenetic clock". Aging. 7 (5): 294–306. doi:10.18632/aging.100742. PMC 4468311. PMID 26000617.
- ↑ Horvath S, Langfelder P, Kwak S, Aaronson J, Rosinski J, Vogt TF, et al. (July 2016). "Huntington's disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels". Aging. 8 (7): 1485–1512. doi:10.18632/aging.101005. PMC 4993344. PMID 27479945.
- ↑ Horvath S, Levine AJ (November 2015). "HIV-1 Infection Accelerates Age According to the Epigenetic Clock". The Journal of Infectious Diseases. 212 (10): 1563–1573. doi:10.1093/infdis/jiv277. PMC 4621253. PMID 25969563.
- ↑ Gross AM, Jaeger PA, Kreisberg JF, Licon K, Jepsen KL, Khosroheidari M, et al. (April 2016). "Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA". Molecular Cell. 62 (2): 157–168. doi:10.1016/j.molcel.2016.03.019. PMC 4995115. PMID 27105112.
- ↑ Horvath S, Ritz BR (December 2015). "Increased epigenetic age and granulocyte counts in the blood of Parkinson's disease patients". Aging. 7 (12): 1130–1142. doi:10.18632/aging.100859. PMC 4712337. PMID 26655927.
- ↑ Walker RF, Liu JS, Peters BA, Ritz BR, Wu T, Ophoff RA, Horvath S (May 2015). "Epigenetic age analysis of children who seem to evade aging". Aging. 7 (5): 334–339. doi:10.18632/aging.100744. PMC 4468314. PMID 25991677.
- 1 2 3 4 Levine ME, Lu AT, Chen BH, Hernandez DG, Singleton AB, Ferrucci L, et al. (August 2016). "Menopause accelerates biological aging". Proceedings of the National Academy of Sciences of the United States of America. 113 (33): 9327–9332. doi:10.1073/pnas.1604558113. PMC 4995944. PMID 27457926.
- 1 2 3 Lowe D, Horvath S, Raj K (February 2016). "Epigenetic clock analyses of cellular senescence and ageing". Oncotarget. 7 (8): 8524–8531. doi:10.18632/oncotarget.7383. PMC 4890984. PMID 26885756.
- ↑ Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. (November 2011). "Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders". Nature. 479 (7372): 232–236. Bibcode:2011Natur.479..232B. doi:10.1038/nature10600. PMC 3468323. PMID 22048312.
- 1 2 3 Horvath S, Gurven M, Levine ME, Trumble BC, Kaplan H, Allayee H, et al. (August 2016). "An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease". Genome Biology. 17 (1): 171. doi:10.1186/s13059-016-1030-0. PMC 4980791. PMID 27511193.
- ↑ Stölzel F, Brosch M, Horvath S, Kramer M, Thiede C, von Bonin M, et al. (August 2017). "Dynamics of epigenetic age following hematopoietic stem cell transplantation". Haematologica. 102 (8): e321–e323. doi:10.3324/haematol.2016.160481. PMC 5541887. PMID 28550187.
- ↑ Maierhofer A, Flunkert J, Oshima J, Martin GM, Haaf T, Horvath S (April 2017). "Accelerated epigenetic aging in Werner syndrome". Aging. 9 (4): 1143–1152. doi:10.18632/aging.101217. PMC 5425119. PMID 28377537.
- ↑ Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, et al. (July 2018). "Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies". Aging. 10 (7): 1758–1775. doi:10.18632/aging.101508. PMC 6075434. PMID 30048243.
- ↑ Fleischer JG, Schulte R, Tsai HH, Tyagi S, Ibarra A, Shokhirev MN, Huang L, Hetzer MW, Navlakha S (December 2018). "Predicting age from the transcriptome of human dermal fibroblasts". Genome Biology. 19 (1): 221. doi:10.1186/s13059-018-1599-6. PMC 6300908. PMID 30567591.
- ↑ Vilenchik MM, Knudson AG (October 2003). "Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer". Proceedings of the National Academy of Sciences of the United States of America. 100 (22): 12871–12876. Bibcode:2003PNAS..10012871V. doi:10.1073/pnas.2135498100. PMC 240711. PMID 14566050.
- ↑ Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, et al. (July 2007). "DNA damage, homology-directed repair, and DNA methylation". PLOS Genetics. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- ↑ O'Hagan HM, Mohammad HP, Baylin SB (August 2008). "Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island". PLOS Genetics. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- ↑ Morano A, Angrisano T, Russo G, Landi R, Pezone A, Bartollino S, et al. (January 2014). "Targeted DNA methylation by homology-directed repair in mammalian cells. Transcription reshapes methylation on the repaired gene". Nucleic Acids Research. 42 (2): 804–821. doi:10.1093/nar/gkt920. PMC 3902918. PMID 24137009.
- ↑ Ding N, Bonham EM, Hannon BE, Amick TR, Baylin SB, O'Hagan HM (June 2016). "Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage". Journal of Molecular Cell Biology. 8 (3): 244–254. doi:10.1093/jmcb/mjv050. PMC 4937888. PMID 26186941.
- 1 2 Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, et al. (February 2014). "Aging of blood can be tracked by DNA methylation changes at just three CpG sites". Genome Biology. 15 (2): R24. doi:10.1186/gb-2014-15-2-r24. PMC 4053864. PMID 24490752.
- ↑ Horvath S (2014-02-18 16:34) Comparison with the epigenetic clock (2014). Reader Comment.
- ↑ Wagner W (2014) Response to comment "comparison with the epigenetic clock by Horvath 2013"
- ↑ "Horvath S (2013-11-04 11:00) Erratum in cancer tissues Reader Comment". Archived from the original on 2014-04-13. Retrieved 2014-04-14.
- ↑ Giuliani C, Cilli E, Bacalini MG, Pirazzini C, Sazzini M, Gruppioni G, et al. (April 2016). "Inferring chronological age from DNA methylation patterns of human teeth". American Journal of Physical Anthropology. 159 (4): 585–595. doi:10.1002/ajpa.22921. PMID 26667772.
- ↑ Galkin, F.; Mamoshina, P.; Kochetov, K.; Sidorenko, D.; Zhavoronkov, A. (2020). "DeepMAge: A Methylation Aging Clock Developed with Deep Learning". Aging and Disease. doi:10.14336/AD.
- ↑ Bock, Christoph; et al. (July 2016). "Quantitative comparison of DNA methylation assays for biomarker development and clinical applications". Nature Biotechnology. 34 (7): 726–737. doi:10.1038/nbt.3605. hdl:10261/173638. PMID 27347756. S2CID 205283232.
- ↑ de Lima Camillo, Lucas Paulo; Lapierre, Louis R.; Singh, Ritambhara (2022-04-19). "A pan-tissue DNA-methylation epigenetic clock based on deep learning". npj Aging. 8 (1): 1–15. doi:10.1038/s41514-022-00085-y. ISSN 2731-6068. PMC 9158789. S2CID 248270807.
- ↑ AltumAge, Singh Lab @ Brown, 2022-04-19, retrieved 2022-04-21
- ↑ Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, et al. (March 2017). "Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment". Genome Biology. 18 (1): 57. doi:10.1186/s13059-017-1186-2. PMC 5371228. PMID 28351423.
- ↑ Petkovich DA, Podolskiy DI, Lobanov AV, Lee SG, Miller RA, Gladyshev VN (April 2017). "Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions". Cell Metabolism. 25 (4): 954–960.e6. doi:10.1016/j.cmet.2017.03.016. PMC 5578459. PMID 28380383.
- ↑ Stubbs TM, Bonder MJ, Stark AK, Krueger F, von Meyenn F, Stegle O, Reik W (April 2017). "Multi-tissue DNA methylation age predictor in mouse". Genome Biology. 18 (1): 68. doi:10.1186/s13059-017-1203-5. PMC 5389178. PMID 28399939.
- ↑ Wagner W (June 2017). "Epigenetic aging clocks in mice and men". Genome Biology. 18 (1): 107. doi:10.1186/s13059-017-1245-8. PMC 5470213. PMID 28615041.
- ↑ Fairfield EA, Richardson DS, Daniels CL, Butler CL, Bell E, Taylor MI (September 2021). "Ageing European lobsters (Homarus gammarus) using DNA methylation of evolutionarily conserved ribosomal DNA". Evolutionary Applications. 14 (9): 2305–2318. doi:10.1111/eva.13296. PMC 8477595. PMID 34603500.
- ↑ De Paoli-Iseppi R, Deagle BE, McMahon CR, Hindell MA, Dickinson JL, Jarman SN (17 August 2017). "Measuring Animal Age with DNA Methylation: From Humans to Wild Animals". Frontiers in Genetics. 8: 106. doi:10.3389/fgene.2017.00106. PMC 5572392. PMID 28878806.
- ↑ Amin Haghani; et al. (2023). "DNA methylation networks underlying mammalian traits". Science. 381 (6658): eabq5693. doi:10.1126/science.abq5693. hdl:20.500.11820/eaafb00f-2c73-44ba-91ec-bc9b304f0bdd.
Further reading
- Simpson DJ, Chandra T (September 2021). "Epigenetic age prediction". Aging Cell. 20 (9): e13452. doi:10.1111/acel.13452. PMC 8441394. PMID 34415665.
- Aquino E, Benton M, Haupt L, Sutherland H, Griffiths L, Lea R (12 April 2018). "Current Understanding of DNA Methylation and Age-related Disease". OBM Genetics. 2 (2): 1. doi:10.21926/obm.genet.1802016.
- Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD (September 2018). "DNA Methylation Clocks in Aging: Categories, Causes, and Consequences". Molecular Cell. 71 (6): 882–895. doi:10.1016/j.molcel.2018.08.008. PMC 6520108. PMID 30241605.
- Bell CG, Lowe R, Adams PD, Baccarelli AA, Beck S, Bell JT, et al. (November 2019). "DNA methylation aging clocks: challenges and recommendations". Genome Biology. 20 (1): 249. doi:10.1186/s13059-019-1824-y. PMC 6876109. PMID 31767039.
- Wang M, Lemos B (March 2019). "Ribosomal DNA harbors an evolutionarily conserved clock of biological aging". Genome Research. 29 (3): 325–333. doi:10.1101/gr.241745.118. PMC 6396418. PMID 30765617.
- Bergsma T, Rogaeva E (2020). "DNA Methylation Clocks and Their Predictive Capacity for Aging Phenotypes and Healthspan". Neuroscience Insights. 15: 2633105520942221. doi:10.1177/2633105520942221. PMC 7376380. PMID 32743556.
External links
- Wickelgren, Ingrid (2022-08-17). "Epigenetic 'Clocks' Predict Animals' True Biological Age". Quanta Magazine.